
Introduction
The immune system is a complex network of biological components and processes that maintains homeostasis by defending the body against external pathogens, including viruses, bacteria, and parasites (Alavi et al., 2020; Ji et al., 2021a; Park et al., 2023). As human life expectancy rises, the decline in immune function with age has become an increasingly important public health concern (Montecino-Rodriguez et al., 2013). Additionally, modern lifestyles, dietary habits, and environmental hazards can negatively impact immunity, further underscoring the need to maintain a healthy immune system (Kim et al., 2022). This has fueled growing interest in natural products with immunostimulatory properties (Cheng et al., 2019; Zhao et al., 2020).
Polysaccharides have attracted significant attention for their immunomodulatory effects, and extensive research has been conducted in this area (Chen & Huang, 2018; Huang et al., 2019; Li et al., 2022; Yu et al., 2018). For instance, mushroom-derived polysaccharides such as β-glucan have been shown to enhance immune function through various biological activities, including immunomodulation, anticancer effects, and anti-inflammatory properties (Chakraborty et al., 2021; Maity et al., 2021; Zhao et al., 2020). Fucoidan, a complex sulfated polysaccharide obtained from the cell walls of brown seaweeds, is another compound known for its broad spectrum of biological activities, particularly its ability to boost immune function (Apostolova et al., 2020; Li et al., 2008; Wijesinghe & Jeon, 2012; Yang et al., 2023).
Most research on fucoidan has focused on its extraction from Undaria pinnatifida, and this fucoidan has been utilized in preventing and treating diseases, as well as in functional foods, materials, and pharmaceuticals (Bi et al., 2018; Zeng et al., 2022; Zhao et al., 2018). However, the structure and bioactivity of fucoidan can be subject to variation depending on the algal source from which it is extracted (Cumashi et al., 2007). Sargassum fusiforme, another species of brown seaweed, has long been valued for its edible and medicinal uses (Li et al., 2006). Studies have demonstrated that S. fusiforme can modulate immune function in immune cells, such as macrophages and splenocytes (Shan et al., 1999; Yoon et al., 2011). However, the immunomodulatory properties of fucoidan extracted specifically from S. fusiforme have not been sufficiently explored.
Thus, this study aims to assess the immunostimulatory properties of S. fusiforme fucoidan (SFF) using murine macrophages, specifically RAW 264.7 cells. To achieve this, we examined nitric oxide (NO) and cytokine production, phagocytic activity, the expression of inducible NO synthase (iNOS) and cyclooxygenase 2 (COX-2), and the activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways.
Materials and Methods
Lipopolysaccharide (LPS) and 4′,6-Diamidino-2-phenylindole (DAPI) were obtained from Sigma-Aldrich Chemical (St. Louis, MO, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and NE-PER Nuclear/Cytoplasmic Extraction Reagents were purchased from Invitrogen (Carlsbad, CA, USA) and Thermo Fisher Scientific (Waltham, MA, USA), respectively. The phagocytosis assay kit (catalog No. 500290) was bought from Cayman Chemical (Ann Arbor, MI, USA). ELISA kits for interleukin (IL)-1β (catalog No. MLB00C), IL-6 (catalog No. M6000B), and tumor necrosis factor-α (TNF-α, catalog No. MTA00B) were acquired from R&D Systems (Minneapolis, MN, USA).
S. fusiforme (SF) was procured from Sinchon, Jeju City, Korea. The SF was rinsed three times with running water to remove impurities and salt, followed by far-infrared drying and crushing. To further eliminate impurities such as ash and salt, the crushed SF was soaked in water at a volume 50 times its weight for 6 h, after which the supernatant was decanted (Fig. 1). An alkali pretreatment was then performed by adding 1% sodium hydrogen carbonate (at 50 times the volume of the sample) and incubating the mixture in a bath at 75°C for 2 h.

For enzyme extraction, a solution with 15% solvent and 5% enzyme (relative to the pretreated SF) was added, and the mixture was incubated in a shaking incubator at 50°C for 24 h. The enzyme was deactivated by heating the post-extraction material in the bath at 100°C, followed by separation of residual material via centrifugation. To precipitate alginate, a 6 M CaCl2 solution (15% of the supernatant volume) was added, and the mixture was incubated at 4°C for 8–12 h. The precipitated alginate was then removed through centrifugation. The remaining supernatant was desalinated using a 12 kDa membrane filter until the salinity reached 30 ppm or lower. The resulting crude fucoidan extract was concentrated to 20% of the total volume and freeze-dried. The final crude fucoidan (SFF) contained 52.5 ± 0.5% polysaccharides and 20.5 ± 0.5% sulfate.
Murine macrophage RAW 264.7 cells were provided by the Korea Cell Line Bank (Seoul, Korea) and grown in Dulbecco’s Modified Eagle’s Medium (DMEM; WelGENE, Daegu, Korea) enriched with 1% penicillin-streptomycin (catalog No. 15140-122, Gibco BRL, Grand Island, NY, USA) and 10% fetal bovine serum (FBS; Gibco BRL). The cells were grown until they reached 80%–90% confluency and incubated at 37°C with 5% CO2.
RAW 264.7 macrophages were cultured at a density of 5 × 10⁵ cells per well in 6-well plates. After being maintained for 24 h, the cells were exposed to varying concentrations of SFF (25, 50, and 100 μg/mL) or 10 ng/mL LPS. To assess cell viability via the MTT assay, a 500 μg/mL MTT solution was introduced to each well. After allowing for 2 h of formazan crystal formation, the products were solubilized in dimethyl sulfoxide (DMSO; catalog No. 33626, Sigma-Aldrich Chemical), and absorbance values of each sample were obtained at 540 nm with a microplate reader (VERSA Max, Molecular Devices, Sunnyvale, CA, USA) (Kwon et al., 2019a). Using an inverted phase-contrast microscope at 400× magnification (Carl Zeiss, Oberkochen, Germany), morphological changes in the SFF- or LPS-treated cells were visualized.
NO production by macrophages was determined colorimetrically via the Griess reaction. The Griess reagent was formulated by combining equal proportions of Griess reagent A (1% sulfanilamide in 5% phosphoric acid) (catalog Nos. 33626; 49685, Sigma-Aldrich Chemical) and Griess reagent B (0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in distilled water) (catalog No. 33461, Sigma-Aldrich Chemical). The Griess reagent was introduced to the cell culture supernatant at an equal volume and maintained in the dark for 20 min. Absorbance was then determined at a wavelength of 540 nm. NO concentration was interpolated from a standard calibration curve generated with sodium nitrite (NaNO2; catalog No. 7632-00-0, Sigma-Aldrich Chemical) (Lee et al., 2017).
Phagocytic activity was evaluated by employing a commercial phagocytosis assay kit (Kwon et al., 2019b). RAW 264.7 cells were cultured in 6-well plates with a cell seeding density of 5 × 10⁵ cells per well. After being maintained for 24 h, Various concentrations of SFF (25, 50, and 100 μg/mL) or 10 ng/mL LPS were applied to the cells followed by incubation for an additional 24 h. Fluorescently labeled latex beads conjugated to rabbit IgG and fluorescein isothiocyanate (FITC) were added to each well at a dilution of 1:1,000. Following 2 h of incubation, the cells underwent two washes with phosphate-buffered saline (PBS) and were stained with 40 μM DAPI to visualize the cell nuclei. Following a 10 min incubation, fluorescence intensity was analyzed by flow cytometry (BD FACSVerse, BD Biosciences, San Jose, CA) and fluorescence microscopy (Leica Microsystems, Wentzler, Germany).
RAW 264.7 cells were cultured in 6-well plates with a cell seeding density of 5 × 10⁵ cells per well. After being maintained for 24 h, the cells were exposed to various concentrations SFF (25, 50, and 100 μg/mL) or 10 ng/mL LPS for another 24 h. The supernatants were harvested and determined the concentrations of TNF-α, IL-6, and IL-1β employing ELISA kits as instructed by the manufacturer.
RAW 264.7 cells were exposed to varying concentrations of SFF or LPS. After incubation, the cells were harvested and washed twice with ice-cold PBS. Total protein was extracted using a cell lysis buffer. Additionally, nuclear and cytoplasmic protein fractions were isolated using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents kit, following the manufacturer’s instructions. The Bio-Rad Protein Assay kit (Bio‐Rad Laboratories, Hercules, CA, USA) was utilized to quantify protein concentrations. Protein samples were heat-denatured for 10 min before performing SDS-PAGE to ensure equal loading. The separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Bedford, MA, USA). Nonspecific binding was prevented by blocking the membranes with 5% skim milk in PBS containing 0.1% Tween 20 (PBST) for 1 h at room temperature, after which the membranes were probed with specific primary antibodies overnight at 4°C. Following the wash with PBST to remove unbound antibodies, the membranes were treated with secondary antibodies for 1 h at room temperature. Protein detection and analysis were performed using the Fusion FX Image system (Vilber Lourmat, Torcy, France), and data were quantified using ImageJ® software (v1.48, NIH, Bethesda, MD, USA). The following primary antibodies were used: β-actin (BS6007M, Bioworld Technology, MN, USA), Lamin B (sc-6216, Santa Cruz Biotechnology, Paso Robles, CA, USA), iNOS (610333, BD Biosciences), COX-2 (12282S, Cell Signaling Technology, Beverly, MA, USA), Inhibitor of kappa B alpha (IκB-α; SC371, Santa Cruz Biotechnology), NF-κB p65 (6956P, Cell Signaling Technology), Extracellular signal-regulated kinase (ERK; SC1647, Santa Cruz Biotechnology), c-Jun N-terminal kinase (JNK; 9252S, Cell Signaling Technology), p38 (SC7972, Cell Signaling Technology), phospho-ERK (9106S, Cell Signaling Technology), phospho-JNK (9255, Cell Signaling Technology), and phospho-p38 (9211S, Cell Signaling Technology).
All data were expressed as the mean ± SD from three independent experiments. Statistical differences between samples were evaluated using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. These analyses were conducted using GraphPad Prism 10 (GraphPad Software, San Diego, CA, USA).
Results
The cytotoxicity of SFF on RAW 264.7 cells was assessed using the MTT assay. As shown in Fig. 2A, SFF did not present any significant inhibitory effects on cell viability within the concentration range of 25–100 µg/mL, nor did LPS at 10 ng/mL. This indicates that neither SFF nor LPS is cytotoxic to RAW 264.7 cells. Consequently, subsequent experiments were conducted using these treatment concentrations. To assess the impact of SFF on cell activity, NO production was measured. Fig. 2B demonstrates a marked increase in NO production with increasing concentrations of SFF compared to untreated cells.
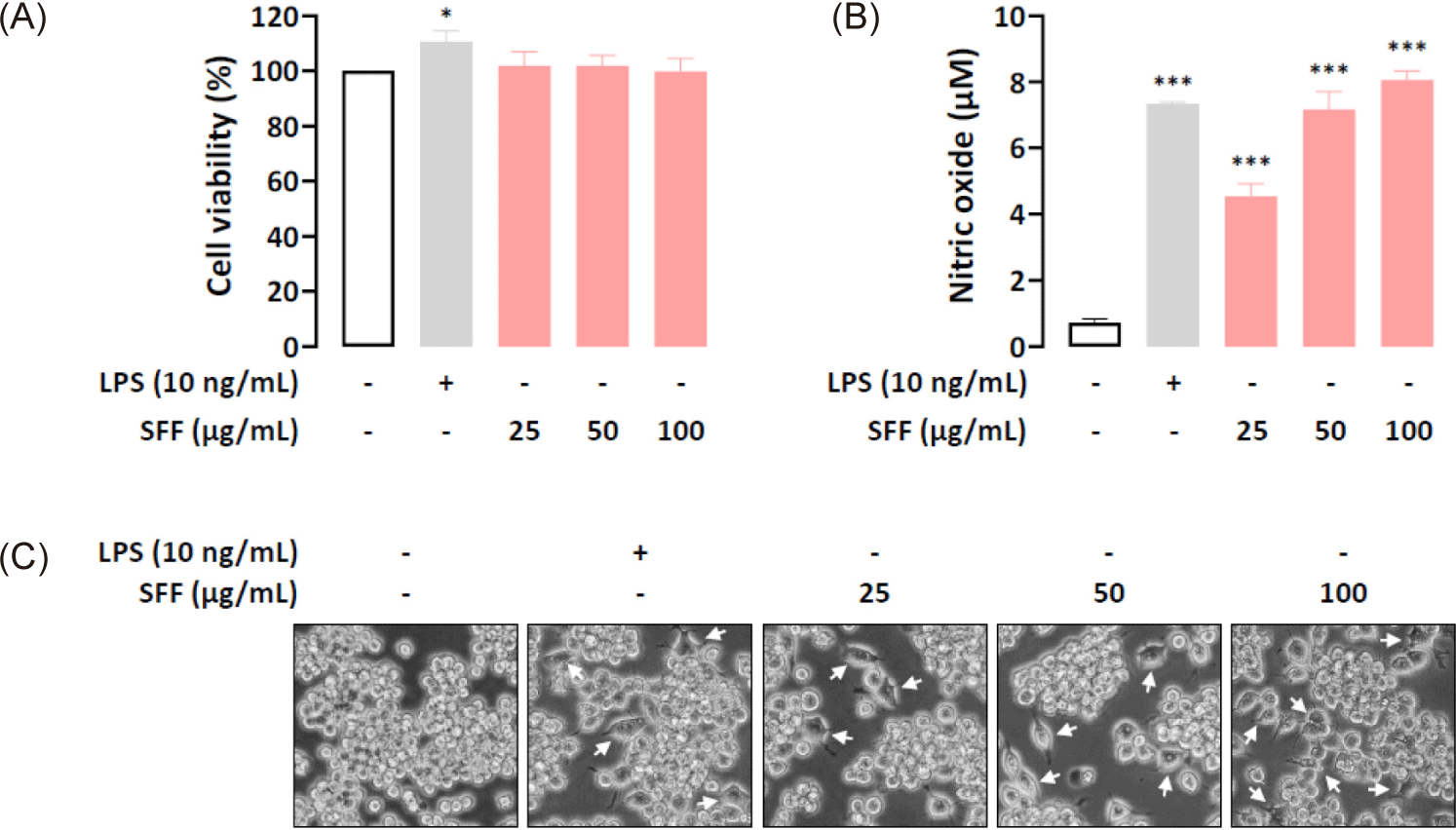
The effects of SFF on cell morphology were evaluated by exposing RAW 264.7 cells to varying concentrations of SFF (25, 50, and 100 µg/mL) or LPS (10 ng/mL). As illustrated in Fig. 2C, cells treated with SFF or LPS exhibited a polygonal spindle shape with elongated pseudopodia, a hallmark of macrophage activation. In contrast, untreated cells retained their round shape and did not display significant morphological changes.
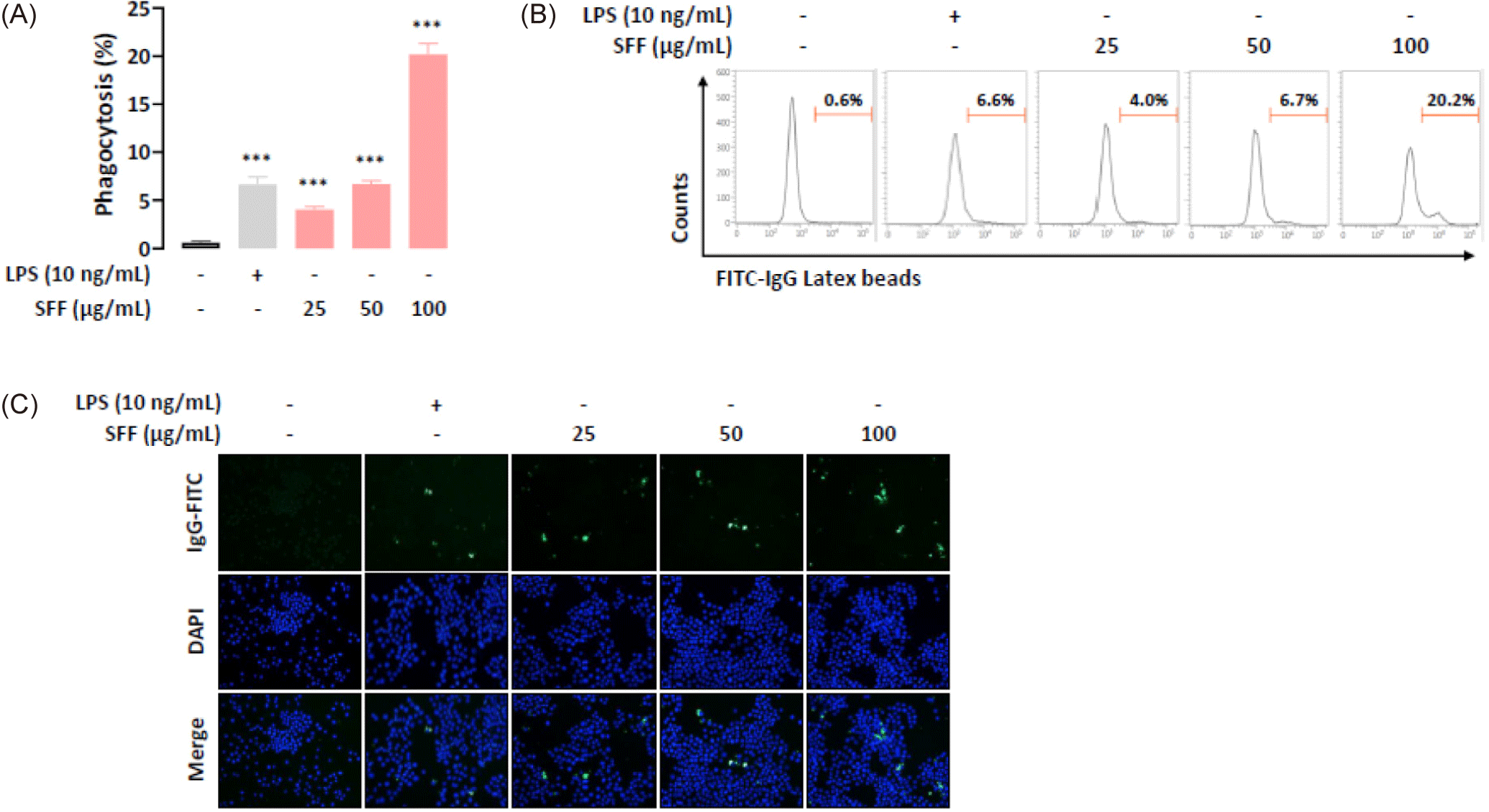
To explore the effect of SFF on phagocytic activity, RAW 264.7 cells were cultured with different concentrations of SFF (25, 50, and 100 µg/mL) or LPS (10 ng/mL). The internalization of FITC-labeled latex beads was quantified using flow cytometry and fluorescence microscopy. Flow cytometry analysis demonstrated a marked increase in phagocytosis with higher concentrations of SFF (Fig. 3A and 3B). Notably, the phagocytic activity observed at 50 µg/mL SFF paralleled that of the positive control group. Phagocytosis increased approximately 30-fold in cells treated with 100 µg/mL SFF. Fluorescence microscopy supported these findings, showing enhanced fluorescence intensity in FITC-IgG-positive cells treated with SFF compared to controls (Fig. 3C). These results indicate that SFF stimulates macrophage activity, consistent with the morphological changes observed in Fig. 2C.
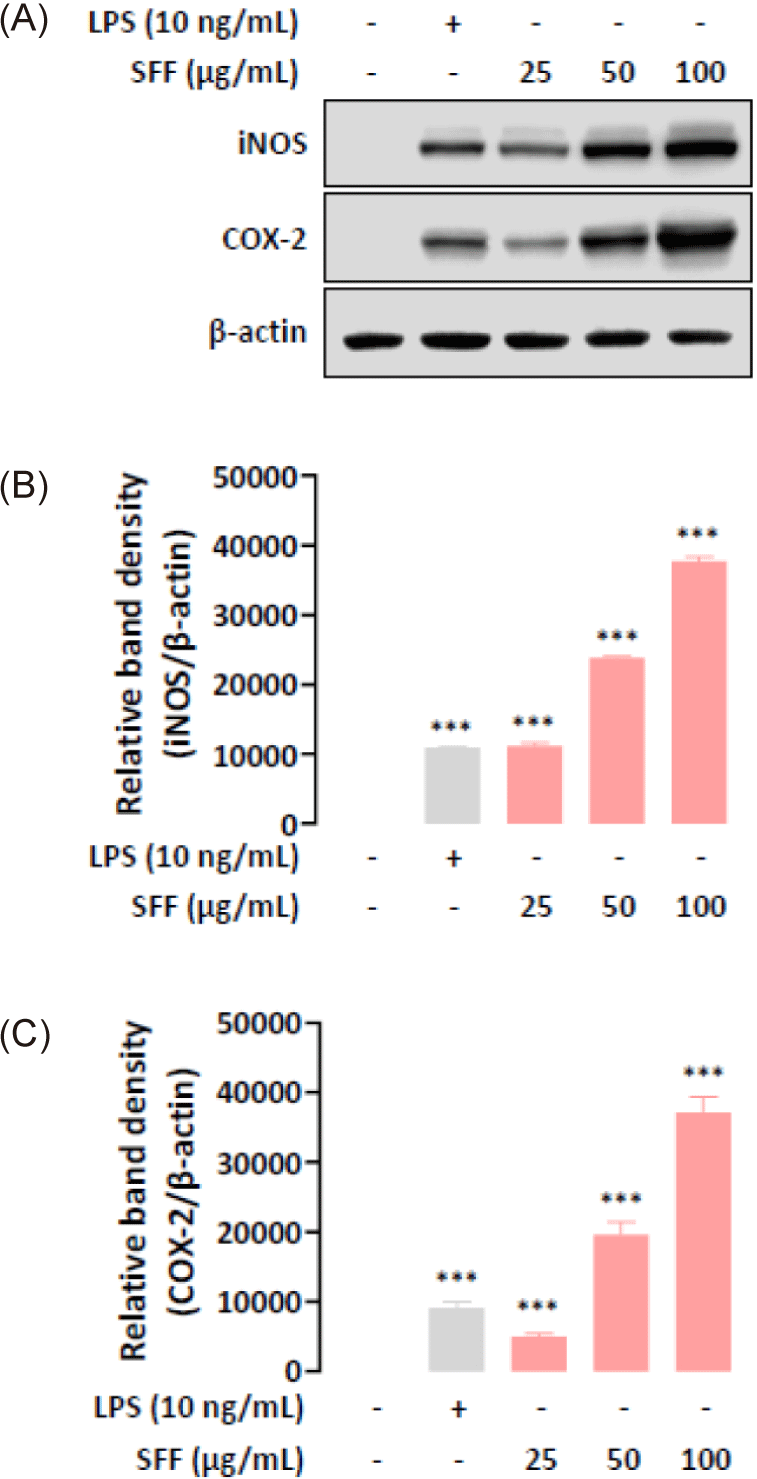
During immune responses, the synthesis of pro-inflammatory mediators NO and prostaglandin E2 (PGE2) is regulated by iNOS and COX-2, respectively (Ji et al., 2021b; Oh et al., 2023). Therefore, we investigated the effects of SFF on iNOS and COX-2 protein expression in macrophages by means of Western blot analysis. As shown in Fig. 4, SFF significantly upregulated iNOS and COX-2 expression dose-dependently.
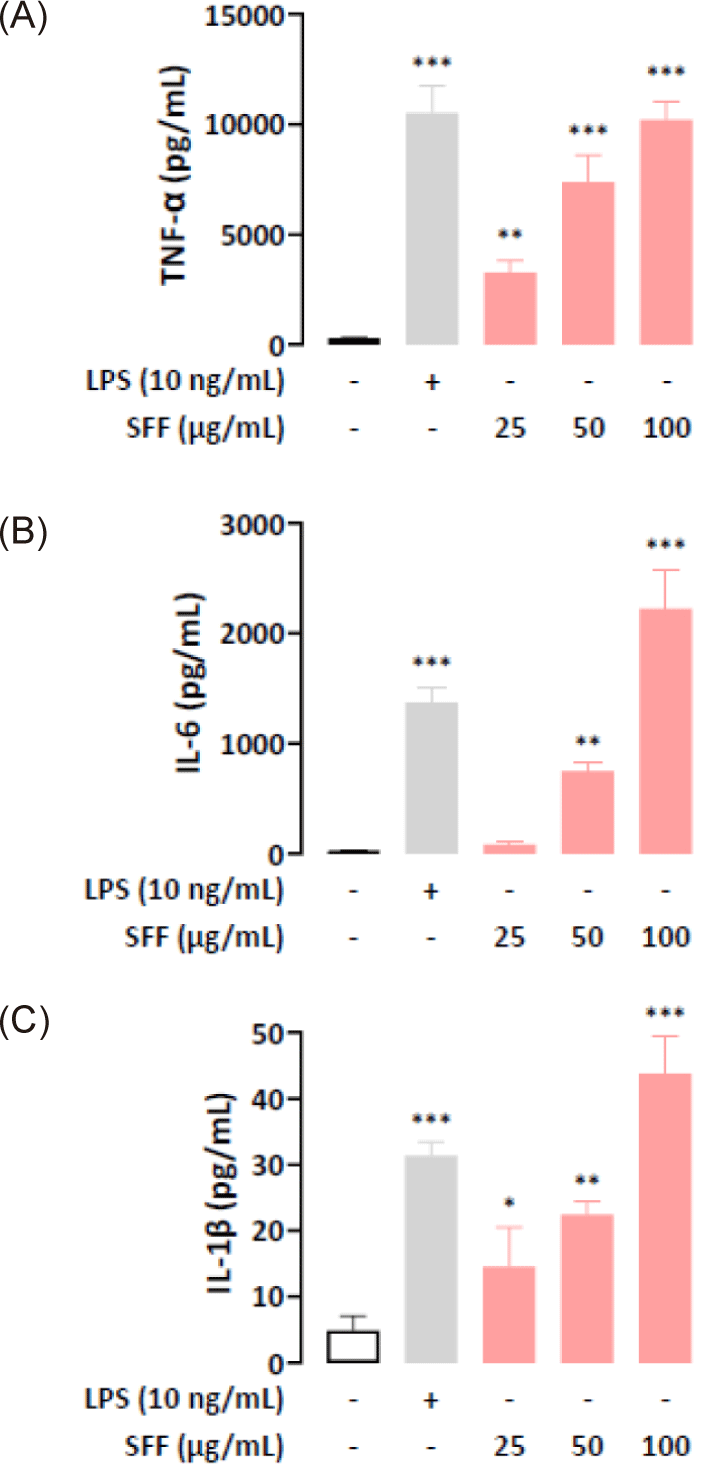
To evaluate cytokine secretion in SFF-treated RAW 264.7 cells, we used ELISA to quantify the production of TNF-α, IL-6, and IL-1β in the cell supernatant. The results indicated that SFF treatment stimulated a dose-dependent increase in cytokine release (Fig. 5). In particular, cytokine levels in cells response to 100 µg/mL SFF were elevated to a similar or greater extent compared to the positive control group.
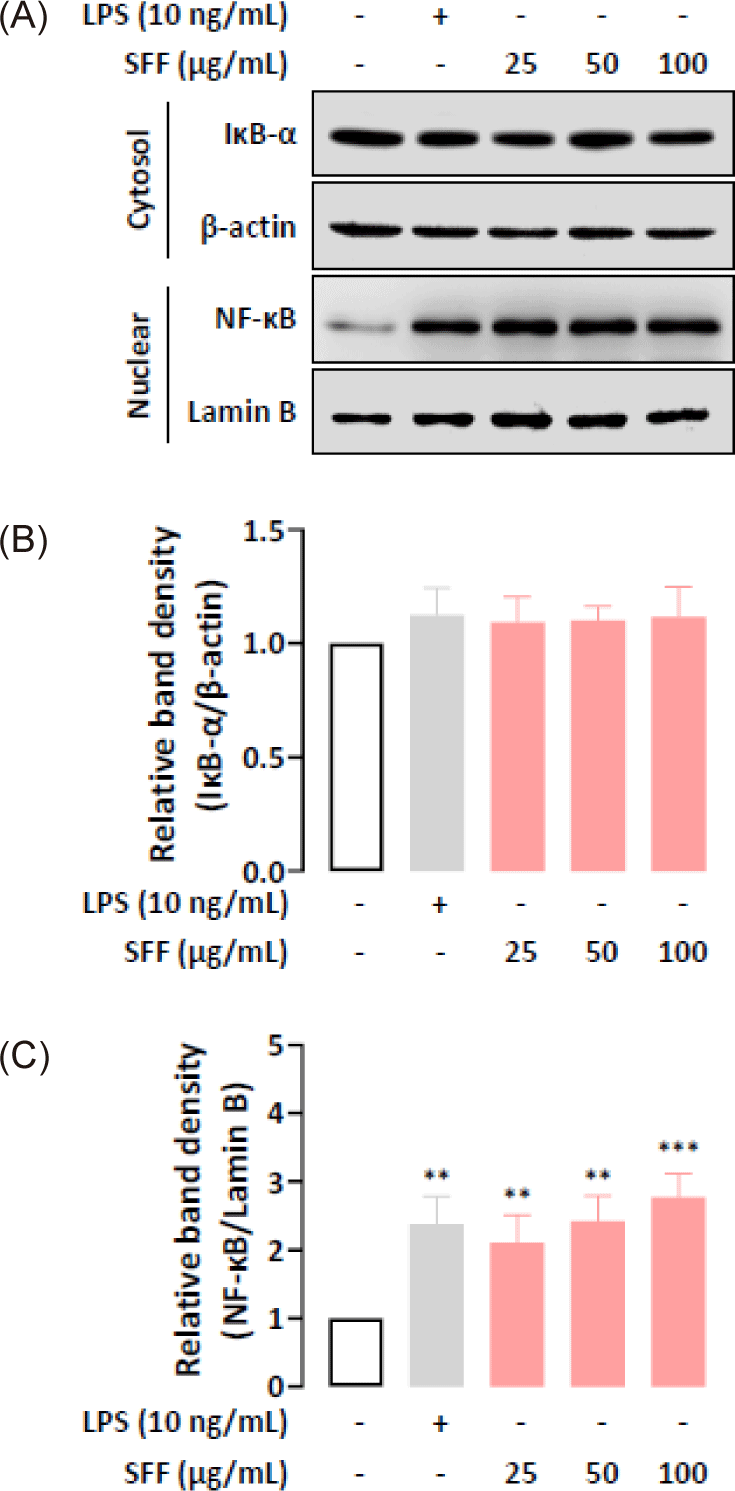
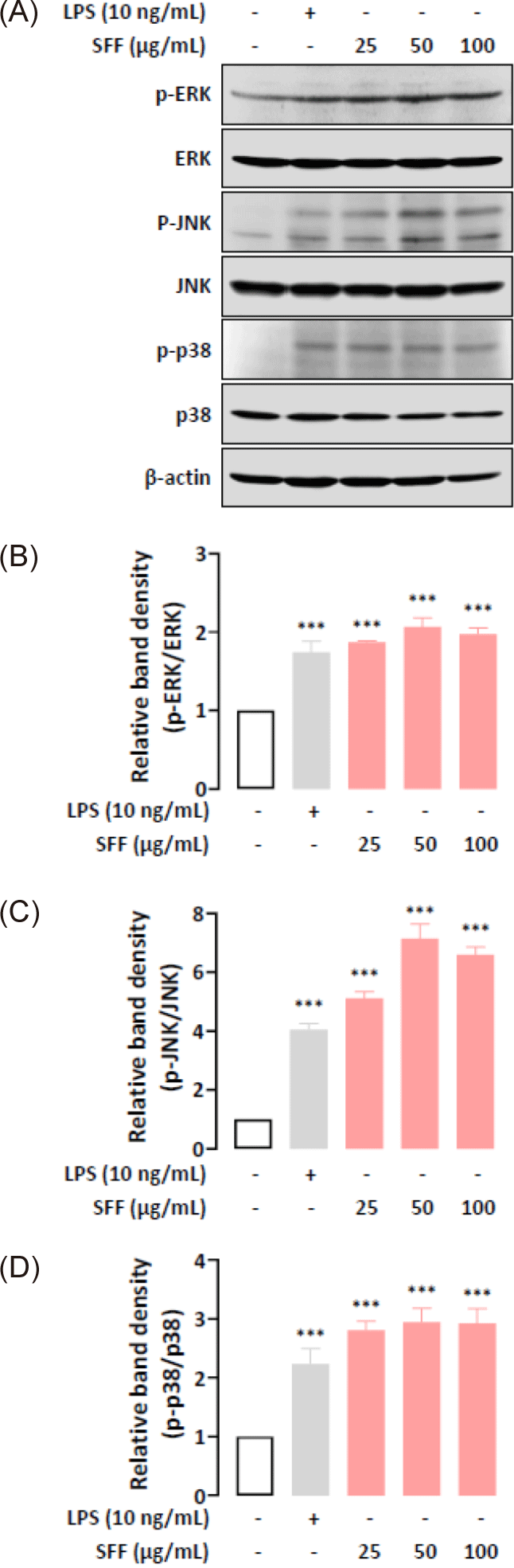
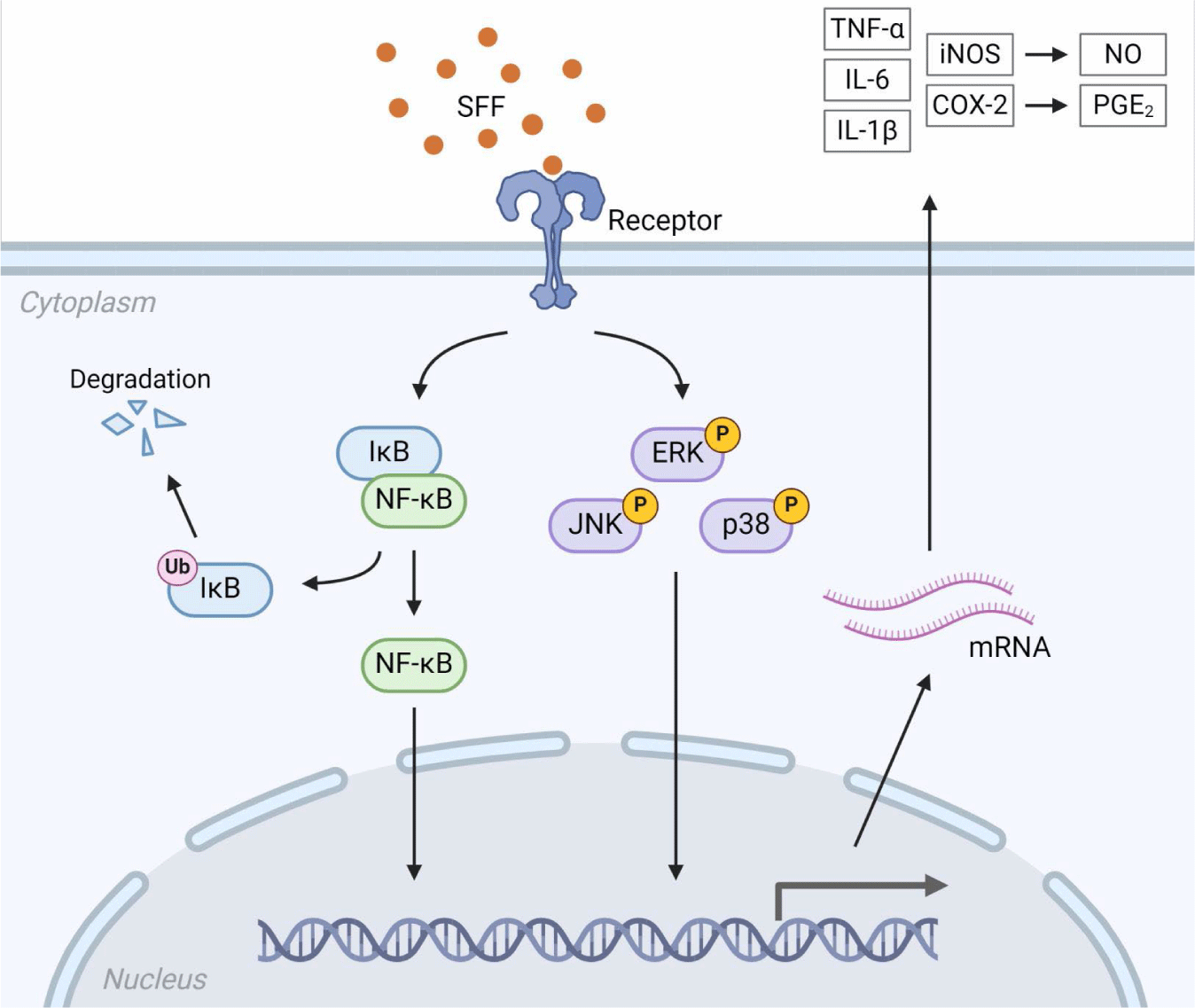
We further examined whether the immunomodulatory functions of SFF were mediated through the NF-κB and MAPK signaling pathways by performing Western blot analysis. Fig. 6 indicate that nuclear NF-κB levels were upregulated in a dose-dependent manner after being treated with SFF for 1 h, which means that NF-κB dissociated from IκB-α in the cytoplasm and relocated to the nucleus. In addition, SFF markedly upregulated the phosphorylation of ERK, JNK, and p38 (Fig. 7), suggesting that the NF-κB and MAPK signaling pathways contribute to the immune-enhancing effects induced by SFF in RAW 264.7 cells (Fig. 8).
Discussion
In this study, the immunomodulatory effects of SFF on RAW 264.7 murine macrophages were investigated. Macrophages, as phagocyte, play a key role in immune defense by increasing phagocytic activity and releasing immune mediators such as NO and cytokines, thereby contributing to both the initiation and maintenance of immune responses (Jiao et al., 2009; Mosser & Edwards, 2008). Enhanced phagocytosis is a hallmark of activated macrophages, enabling them to directly destroy pathogens or tumor cells (Akira et al., 2006). Our findings revealed that SFF treatment significantly boosted phagocytosis, which was accompanied by notable morphological changes in the RAW 264.7 cells.
NO, produced by macrophages, is crucial for killing microbes and tumor cells and serves as an important immune signaling molecule (Ando et al., 2002; Niedbala et al., 2006; Zhou et al., 2017). SFF treatment led to a dose-dependent increase in NO release in RAW 264.7 cells. Western blot result confirmed that SFF upregulated iNOS and COX-2, enzymes responsible for the generation of NO and PGE2, respectively. These results suggest that SFF promotes NO production by upregulating iNOS expression, consistent with previous studies (Cho et al., 2014).
Activated macrophages also regulate immune responses by secreting cytokines such as TNF-α, IL-6, and IL-1β. These cytokines act as signaling molecules to enhance the synthesis of pro-inflammatory mediators, and their production can, in turn, be triggered by these mediators (Apostolova et al., 2020; Kudrin & Ray, 2008). Cytokine production is a widely used indicator of the immunostimulatory effects of biologically active compounds (Guo et al., 2020; Jeong et al., 2015). Our study demonstrated that SFF had a potent stimulatory effect on RAW 264.7 cells, as evidenced by the elevated secretion of these cytokines.
The regulation of pro-inflammatory mediators such as NO, cytokines, iNOS, and COX-2 in activated macrophages is largely controlled by the NF-κB and MAPK signaling pathways (Bi et al., 2018; Khatua et al., 2022; Kim et al., 2022). NF-κB remains inactive in the cytoplasm when bound to IκB-α but translocates to the nucleus upon stimulation, where it induces the transcription of target genes (Doyle & O’Neill, 2006; Hernandez et al., 2019). Our results showed that SFF increased nuclear NF-κB expression levels while maintaining cytoplasmic IκB-α concentrations. Recent studies have indicated that NF-κB can induce an increase in IκB-α synthesis through a self-regulatory feedback mechanism (Thompson et al., 1995). This is attributed to the presence of an NF-κB binding site within the IκB-α promoter. Binding of activated NF-κB to this site induces IκB-α mRNA transcription, resulting in elevated IκB-α protein levels. This establishes a negative feedback loop, whereby the accumulating IκB-α sequesters NF-κB and attenuates its activity. Our results are consistent with this previous research. The MAPK pathway, including ERK, JNK, and p38, is critical for regulating the production of pro-inflammatory mediators (Kaminska, 2005; Oh et al., 2023). The results revealed that SFF stimulates phosphorylation of ERK, JNK, and p38. All of the above findings implied that SFF stimulated the activation of both the NF-κB and MAPK pathways, promoting immune responses and the production of immune mediators in RAW 264.7 cells.
While previous studies have largely focused on the anti-inflammatory effects of fucoidan, this study emphasizes its immunomodulatory role, particularly in the activation of macrophages and the enhancement of immune mediators. This distinction highlights the dual functionality of fucoidan in both mitigating inflammation and regulating broader immune responses. In conclusion, our findings suggest that SFF enhances immunomodulatory activity and offers promising prospects developing functional foods. Further investigations, including in vivo studies, are necessary to elucidate the mechanisms underlying the immunological effects of SFF and to explore the correlation between its structural properties and biological activity.