
Introduction
Muscles consume higher amounts of oxygen than other organs to generate energy for muscle contraction. Therefore, they are vulnerable to oxidative stress, in which free radicals accumulate abnormally and excessively (Di Meo et al., 2017; Steinbacher & Eckl, 2015). Muscle cells are differentiated from myoblasts, which are embryonic precursors, through myogenesis process that fuses into multinucleated myotubes (Abrigo et al., 2019; Sambasivan & Tajbakhsh, 2015). Adequate levels of reactive oxygen species (ROS) can regulate cellular signaling pathways required for muscle differentiation, but excessive ROS accumulation is closely associated with impaired muscle formation. In addition, myoblast apoptosis caused by excessive ROS production is accompanied by cell cycle arrest and DNA damage, contributing to blockage of muscle differentiation and induction of muscle atrophy (Santa-Gonzalez et al., 2016; Yu et al., 2016). Therefore, the level of ROS must be tightly controlled to protect muscle function.
Recently, marine resources are receiving great attention as sources of functional foods and nutraceuticals. Among them, it has been reported that extracts or components of edible seaweed can inhibit oxidative stress-dependent pathological conditions such as inflammation, liver damage, dyslipidemia, endothelial dysfunction, and atherosclerosis (Carroll et al., 2021; Pradhan et al., 2020). Their antioxidant activities mainly involve ROS scavenging and activation of intracellular antioxidant signaling pathways. Several previous results have suggested that nuclear factor-erythroid-2 related factor 2 (Nrf2) is critically involved in the antioxidant efficacy of marine algae extracts or ingredients (Besednova et al., 2022; Jayawardena et al., 2018; Lekshmi et al., 2019; Liu et al., 2021). According to our previous studies, ethanol extracts of marine brown algae Sargassum serratifolium and Petalonia binghamiae can suppress DNA damage and apoptotic cell death in oxidative stress-exposed SW1353 human chondrocytes and mouse-derived C2C12 murine myoblasts by Nrf2-dependent activation of heme oxygenase-1 (HO-1) (Kang et al., 2015; Park et al., 2018). Jayawardena et al. (2018) also demonstrated that activation of Nrf2/HO-1 signaling pathway is mediated by the blocking of fine dust-induced oxidative stress by ethanol extract of S. serratifolium in murine macrophages. In addition, it is reported that various components including seaweed-derived polysaccharides and glycoproteins also exhibit antioxidant activity through a similar mechanism (Lekshmi et al., 2019; Niu et al., 2020).
Myelophycus caespitosus Kjellman 1893, a brown alga belonging to genus Myelophycus, is widely distributed along the coast of Northeastern Asia. It has traditionally been used as a food and medicinal resource (Hanyuda et al., 2020). Although we have reported that a methanol extract of M. caespitosus (MEMC) has potent anti-inflammatory effects in BV2 microglial cells (Jayasooriya et al., 2012), scientific evidence for the pharmacological efficacy of M. caespitosus, including its antioxidant properties, is very limited. Therefore, the objective of the present study was to investigate the antioxidant capacity of MEMC against oxidative stress-mediated cytotoxicity in muscle cells. For this purpose, C2C12 mouse myoblasts were used and treated with hydrogen peroxide (H2O2) to mimic oxidative stress.
Materials and Methods
C2C12 cells (CRL-1772TM, ATCC, Manassas, VA, USA) were cultured using the same method as described previously (Park et al., 2021) and used up until passage number 8. All materials necessary for cell culture were purchased from WELGENE (Gyeongsan, Korea). MEMC provided by Jeju Technopark (Jeju, Korea) and H2O2 (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in dimethyl sulfoxide (Thermo Fisher Scientific, Waltham, MA, USA) to make stock solutions. They were used to treat cells after diluting them to appropriate concentrations with culture medium. C2C12 cells were pretreated with or without MEMC, N-acetyl-L-cysteine (NAC, Thermo Fisher Scientific) or zinc protoporphyrin IX (ZnPP, Sigma-Aldrich) for 1 h before they were treated with H2O2 for 24 h or 1 h.
To investigate cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT, Sigma-Aldrich) assay was applied according to a previously described method (Jeong et al., 2022). Cell morphological changes were observed with an optical microscope (Olympus, Tokyo, Japan).
To investigate cell cycle distribution of cells cultured under different conditions, collected cells were fixed with 70% ethanol. As previously described (Choi, 2022), after staining the cells with propidium iodide (PI, Becton Dickinson, San Jose, CA, USA), the frequency of cells corresponding to each cell cycle was calculated using a flow cytometer (Becton Dickinson). For quantitative evaluation of ROS generation, cells were stained with 10 μM 2’,7’-dichlorofluorescein diacetate (DCF-DA, Becton Dickinson) and then DCF fluorescence intensity reflecting ROS generation was analyzed by flow cytometry. To investigate the mitochondrial membrane potential (MMP) using 5,5’,6,6’-tetrachloro-1,1’3,3’-tetraethyl-imidacarbocyanune iodide (JC-1), cells were stained with 2 μM JC-1 (Thermo Fisher Scientific) and then the percentage of JC-1 monomers using a flow cytometer was expressed to indicate cells that lost MMP. For quantitative evaluation of apoptosis-induced cells, cells were stained with annexin V-fluorescein isothiocyanate and PI (Abcam, Cambridge, UK), and then annexin V-positive cells were considered apoptosis-induced cells as described previously (Choi, 2022).
Immunoblotting was performed using whole, cytoplasmic and mitochondrial proteins extracted as previously described (Mukherjee et al., 2022). Primary and horseradish peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Cell Signaling Technology (Beverly, MA, USA), or Thermo Fisher Scientific. Information on the primary antibodies used in this study is presented in the Table 1. Cytochrome oxidase subunit 4 and actin were used as loading controls for mitochondrial and cytoplasmic proteins.
Cdk, cyclin-dependent kinase; p-γH2AX, phosphorylation of γH2AX; PARP, poly(adenosine diphosphate-ribose) polymerase; Nrf2, nuclear factor-erythroid-2 related factor 2; p-Nrf2, phosphorylation of Nrf2; HO-1, heme oxygenase-1; Keap1, Kelch-like ECH-associated protein-1; COX IV, cytochrome oxidase subunit 4.
Analysis of DNA damage and morphological identification of apoptotic nuclei were performed through Comet assay kit (Trevigen, Gaithersburg, MD, USA) and 4,6-diamidino-2-phenolindole (DAPI, Sigma-Aldrich) staining (Choi, 2022). After comet assay and DAPI staining, fluorescence images were examined and captured by fluorescence microscopy (Carl Zeiss, Oberkochen, Germany) at Core-Facility Center for Tissue Regeneration, Dong-Eui University.
Results
To evaluate the inhibitory activity of MEMC on H2O2-mediated oxidative stress, we assessed investigated the effect of MEMC and H2O2 alone treatment on cell viability of C2C12 cells using the MTT assay. As shown in Fig. 1A, MEMC showed no significant difference in cell viability at concentrations up to 50 mg/mL. Therefore, we used 50 μg/mL of MEMC as the optimal concentration. In H2O2-treated cells, the cell viability was decreased in a dose-dependent manner, and 1 mM H2O2, which reduced cell viability by about half, was set as the cytotoxicity-inducing concentration (Fig. 1B). We next investigated the inhibitory effect of MEMC on H2O2-mediated cytotoxicity and found that MEMC significantly restored H2O2-induced reduction of cell viability and morphological changes (Fig. 1C and 1D), indicating that MEMC pretreatment could improve H2O2-induced cytotoxicity. Furthermore, in the presence of the free radical scavenger NAC, cytotoxicity by H2O2 was completely inhibited (Fig. 1C).
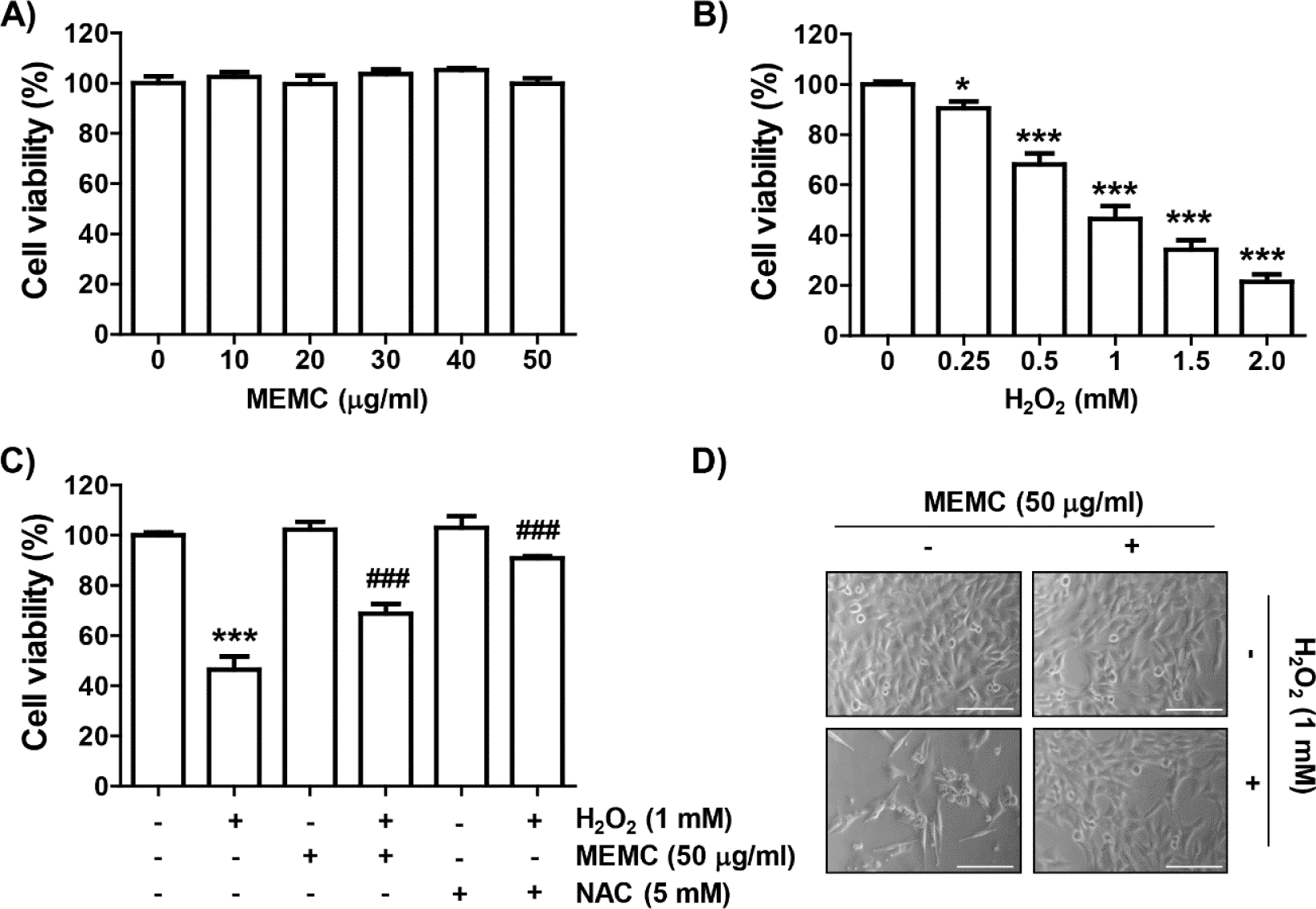
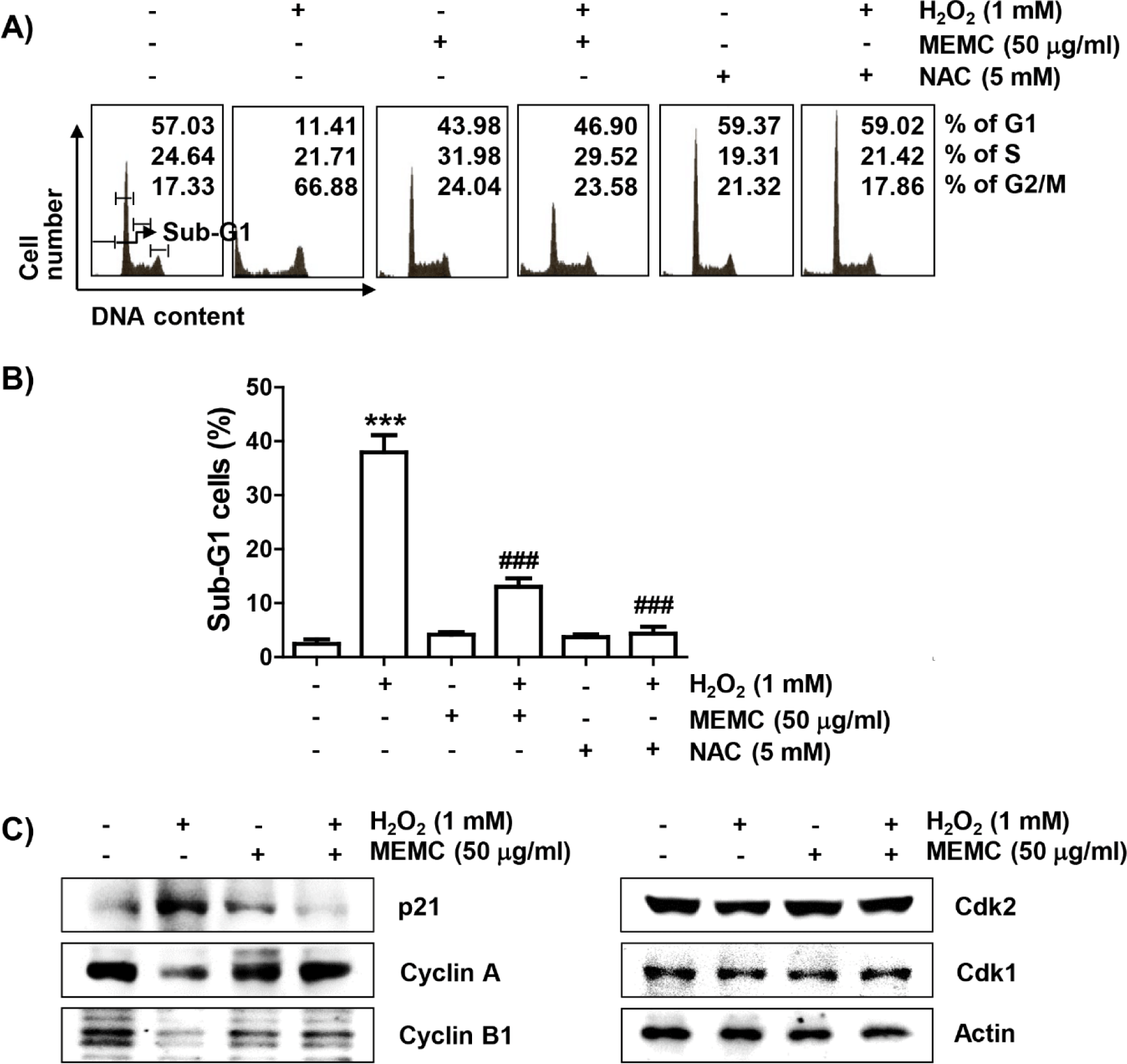
Subsequently, we investigated whether the loss of cell viability in C2C12 cells by H2O2 treatment was due to disturbance of cell cycle progression and induction of apoptosis. As demonstrated in Fig. 2A and 2B, frequencies of cells belonging to G2/M phase and apoptotic sub-G1 phase were increased by H2O2 treatment, but suppressed in the presence of MEMC or NAC. In addition, the expression of p21, protein, one of the representative cyclin-dependent kinase (Cdk) inhibitors, was upregulated in H2O2-treated cells. However, the expression level of cyclin A and cyclin B1 wase suppressed whereas Cdk2 and Cdk1 showed no changes in their expression levels in H2O2-treated cells (Fig. 2C). However, H2O2-induced upregulation of p21 expression was markedly reduced by MEMC treatment, whereas H2O2-induced downregulation of cyclin A and cyclin B1 was inversely increased. These results indicate that the decrease in C2C12 cell survival exposed to H2O2 was due to apoptosis accompanied by cell cycle arrest in the G2/M phase, which was effectively inhibited by MEMC.
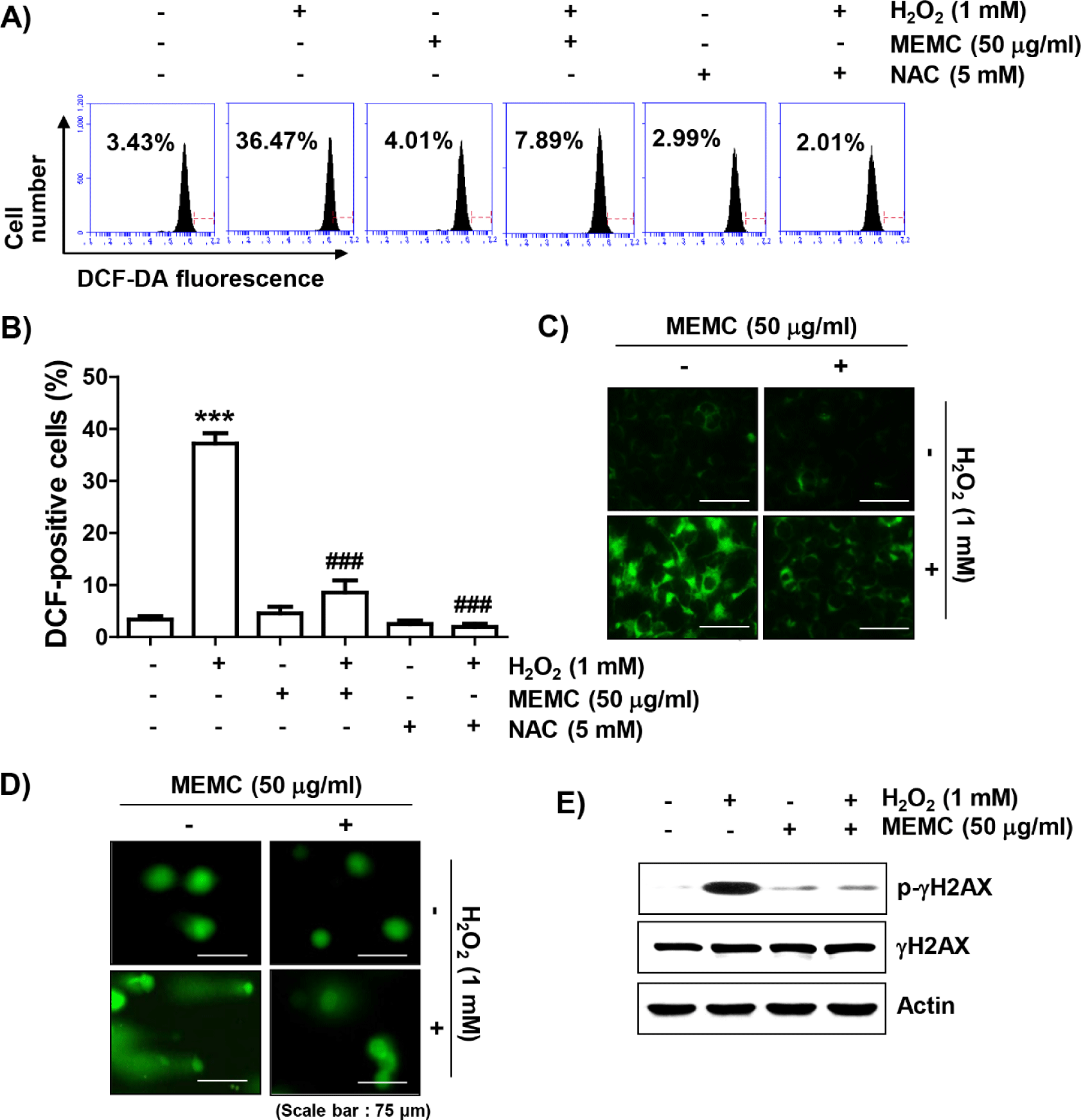
To determine whether the blocking ability of MEMC against H2O2-mediated cytotoxicity was directly related to antioxidant activity, we examined the effect of MEMC on H2O2-induced ROS production using DCF-DA staining. Flow cytometry analysis indicated that ROS production was increased by about 10-fold in C2C12 cells treated with H2O2 for 1 h compared to that in untreated control cells, whereas it was significantly decreased by pretreatment of NAC or MEMC (Fig. 3A, 3B and 3C). Furthermore, in cells treated with H2O2, increases of comet tail moment (DNA migration) and phosphorylation of γH2AX (p-γH2AX) as DNA damage indicators were observed (Fig. 3D and 3E). However, increased DNA migration and phosphorylation of γH2AX by H2O2 treatment were weakened in the presence of MEMC, suggesting that oxidative damage to DNA could be protected by MEMC.
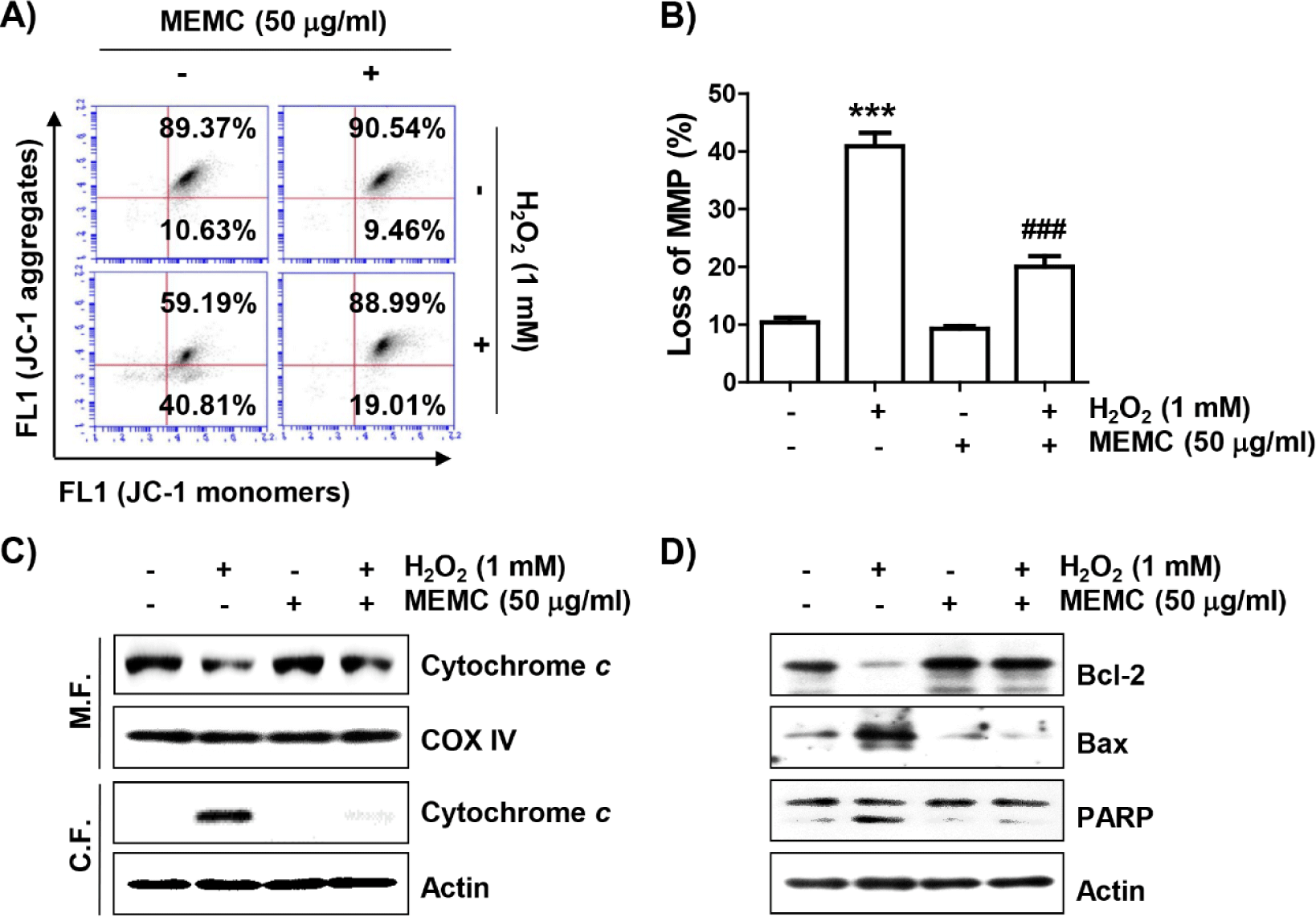
To evaluate whether MEMC could protect against H2O2-induced mitochondrial damage, we estimated MMP following JC-1 staining. JC-1 dye is widely used in apoptosis studies to monitor mitochondrial health, where mitochondria form a monomeric state upon depolarization (Sivandzade et al., 2019). Flow cytometry analysis results (Fig. 4A and 4B) revealed that the frequency of JC-1 monomers was significantly increased whereas that of JC-1 aggregates was decreased in H2O2-treated cells, demonstrating loss of MMP and resultant mitochondrial dysfunction. In addition, after treatment with H2O2, the expression of cytochrome c was increased in the cytoplasmic fraction but decreased in the mitochondrial fraction (Fig. 4C). As shown in Fig. 4D, H2O2 treatment also decreased the expression of Bcl-2 and increased the expression of Bax along with cleavage of poly(adenosine diphosphate-ribose) polymerase (PARP). However, these changes did not occur in cells pretreated with MEMC, suggesting that MEMC was able to inhibit mitochondrial damage caused by H2O2.
To confirm that MEMC prevented H2O2-induced apoptosis, nuclear morphology change observation by DAPI staining and flow cytometry analysis by annexin V/PI staining were performed, respectively. As presented in Fig. 5A and 5B, in H2O2-exposed C2C12 cells, morphological changes such as nuclear fragmentation and chromatin condensation, which are characteristically observed in apoptosis-induced nuclei, were clearly observed. However, MEMC pretreatment markedly attenuated these nuclear morphological changes. In parallel with this, flow cytometry results showed that much more apoptosis was induced in H2O2-treated cells than in control cells (Fig. 5C and 5D). However, the induction of apoptosis by H2O2 was greatly attenuated by MEMC pretreatment, similar to the reduction of sub-G1 cell population.
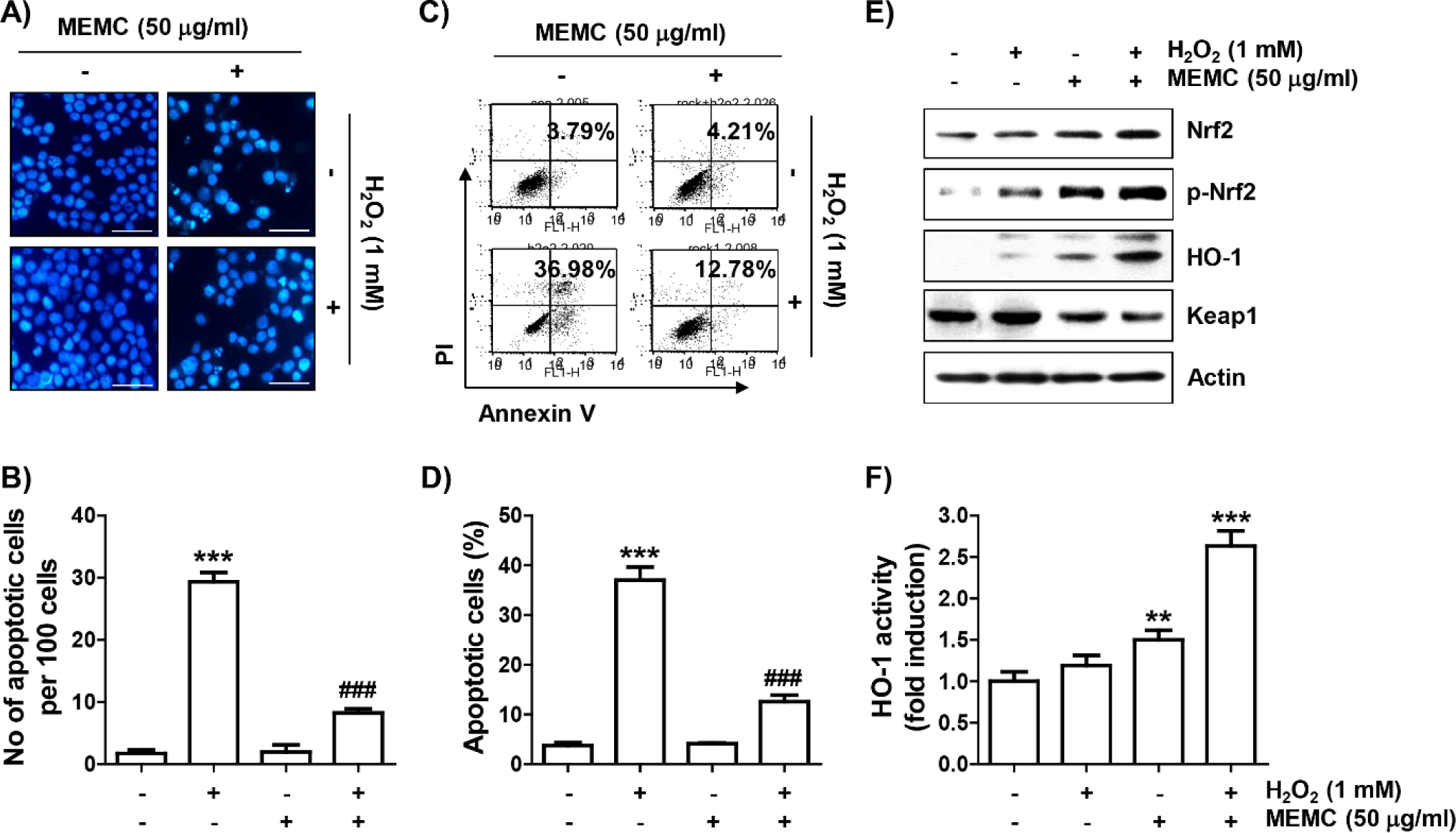
We subsequently investigated whether activation of the Nrf2/HO-1 axis was correlated with the antioxidant capacity of MEMC. As shown in Fig. 5E, the levels of Nrf2 as well as its phosphorylated form (p-Nrf2) were slightly increased by treatment with MEMC or H2O2 alone. However, their expression levels were remarkedly enhanced in H2O2-treated cells after MEMC pretreatment, whereas levels of Kelch-like ECH-associated protein-1 (Keap1) were much lower. Furthermore, the expression and activity of HO-1 were markedly promoted in cells treated with MEMC and H2O2 (Fig. 5E and 5F), suggesting that the Nrf2/HO-1 signaling pathway was further increased by MEMC in H2O2 -treated cells.
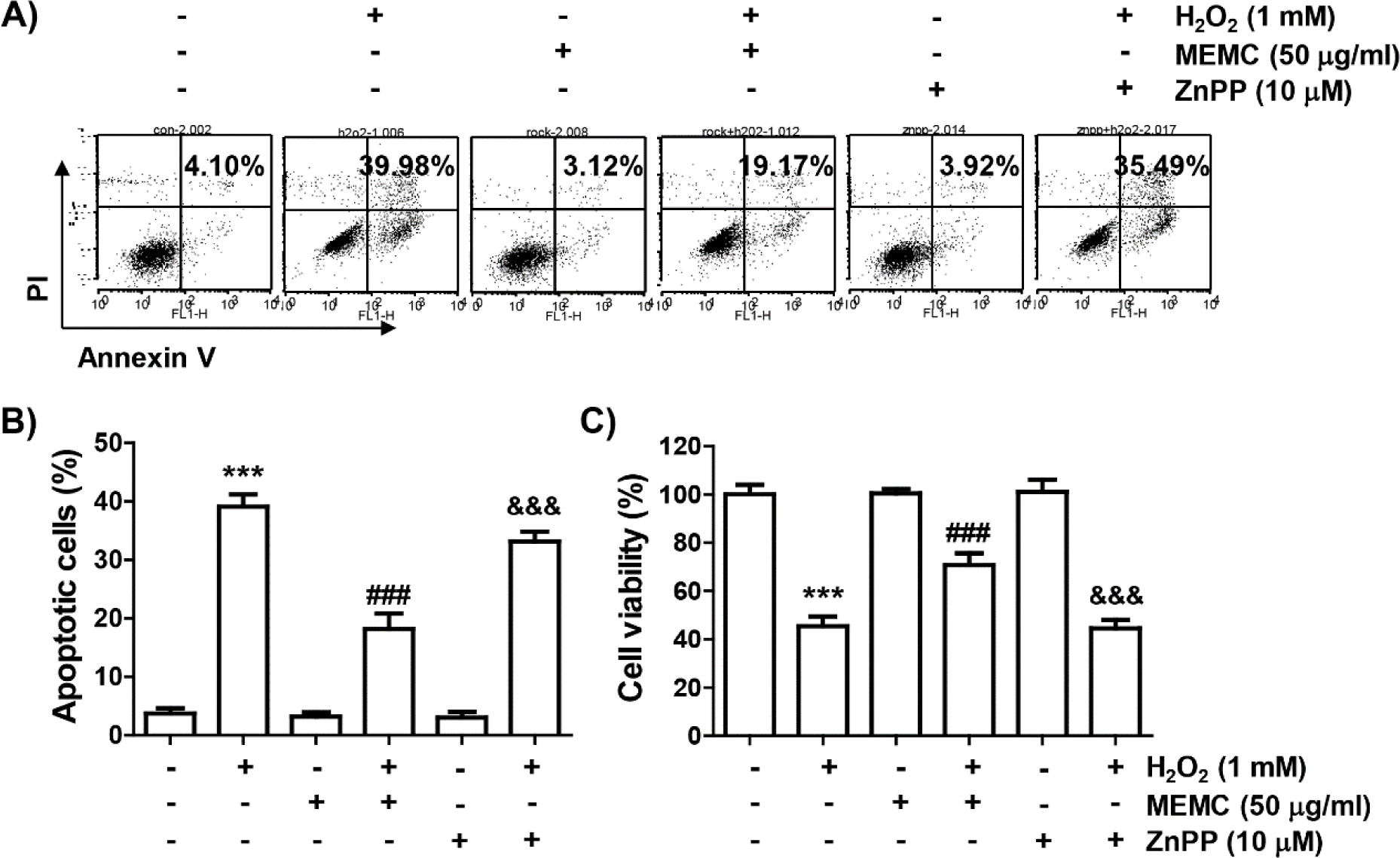
Finally, we used ZnPP, a selective inhibitor of HO-1, to evaluate whether activation of HO-1 by MEMC in H2O2-treated C2C12 cells was directly correlated with the cytotoxicity blocking effect of MEMC. As shown in Fig. 6A, 6B and 6C, the blocking effect of MEMC on apoptosis induced in cells exposed to H2O2 was markedly counteracted in the presence of ZnPP and pretreatment of ZnPP significantly abolished the anti-proliferative effect of MEMC in H2O2-stimulated cells. Consequently, these observations demonstrate that Nrf2-mediated activation of HO-1 at least served as an upstream signal to the inhibitory action of MEMC against H2O2-induced cytotoxicity of C2C12 cells.
Discussion
In the current study, we induced oxidative stress using H2O2 to investigate whether MEMC could protect C2C12 myoblasts from oxidative injury and found that H2O2 treatment induced DNA damage, cell cycle arrest and apoptotic cell death through an increase in ROS generation. However, MEMC was found to be able to block H2O2-induced cytotoxicity while scavenging ROS as an Nrf2 activator.
Induction of cytotoxicity, including DNA damage and apoptosis, by oxidative stimuli is mostly accompanied by cell cycle arrest. Many previous studies have indicated that the cytotoxic effect of H2O2 is closely related to cell cycle arrest at the G2/M phase (Ding et al., 2019; Mubarok et al., 2022). Similar results were also observed in this study using C2C12 myoblasts due to an increase of Cdk inhibitor p21, a negative regulator of cell cycle progression, and decreases of cyclin A and cyclin B1 required for G2 to M phase progression (Dai et al., 2019; Taylor & Stark, 2001). However, H2O2-induced changes of these cell cycle regulators and cell cycle arrest were alleviated by MEMC pretreatment. In addition, the increase in the number of cells corresponding to the sub-G1 stage, which is used as an indicator of apoptosis, was significantly decreased in MEMC-pretreated cells. A similar trend was observed in NAC-pretreated cells. Therefore, it could be predicted that the protective effect of MEMC against H2O2-induced cytotoxicity was associated with inhibition of ROS generation. Similar to NAC pretreatment, MEMC pretreatment completely attenuated the ROS production by H2O2. Moreover, in H2O2-treated C2C12 cells, the formation of comet tails and p-γH2AX, as indicators of oxidative DNA damage (Kopp et al., 2019; Rahmanian et al., 2021), were effectively inhibited by MEMC. Therefore, our results suggest that the ROS scavenging ability of MEMC is responsible for H2O2-induced blockade of cytotoxicity in C2C12 cells.
Apoptosis is largely divided into extrinsic and intrinsic pathways. Overload of ROS by oxidative stress can depolarize mitochondrial membrane and contribute to the activation of the intrinsic apoptosis pathway (Bock & Tait, 2020; Tiwari et al., 2022; Urbani et al., 2021). This results in the loss of MMP, an indicative of mitochondrial dysfunction, and cytosolic release of cytochrome c. It is well known that cytochrome c can activate the caspase cascade required for the mitochondria-mediated intrinsic pathway, resulting in degradation of caspase-dependent proteins such as PARP, thereby terminating apoptosis (Sarwar et al., 2020; Tiwari et al., 2022). In this study, H2O2-induced reduction of MMP, cytosolic release of cytochrome c and cleavage of PARP were largely blocked by MEMC pretreatment. Furthermore, H2O2-induced changes in Bcl-2 family proteins were maintained at control levels after MEMC pretreatment. Accumulated prior studies have shown that the intrinsic pathway is critically controlled by proteins belonging to the Bcl-2 family. Among them, anti-apoptotic proteins including Bcl-2 are essential to maintain stability of the mitochondrial membrane barrier, whereas anti-apoptotic proteins such as Bax are key executors of mitochondrial poration, thereby enhancing mitochondrial membrane permeability and releasing mitochondrial cytochrome c (Bock & Tait, 2020; Tiwari et al., 2022). Several studies have demonstrated that the activation of mitochondria-mediated pathway observed in myoblasts exposed to oxidative stress is typically ROS-dependent (Choi, 2021; Drysch et al., 2021; Yu et al., 2019). In particular, apoptosis, which is observed relatively high in myogenic precursor cells derived from aged muscles, is the cause of weakening of skeletal muscle regeneration, which is related to a high level of ROS (Di Filippo et al., 2016; Fulle et al., 2013; Ji, 2015). These findings well support our data that MEMC can prevent C2C12 myoblasts from apoptosis via suppression of the intrinsic apoptotic pathway by blocking the generation of ROS caused by oxidative stress. Therefore, we propose that treatment with MEMC might contribute to the maintenance of myoblast function and their differentiation into myocytes.
Nrf2 is a redox-sensitive transcription factor that can enhance antioxidant capacity by regulating the expression of phase II detoxification enzymes (Jenkins & Gouge, 2021; Tonelli et al., 2018). When cells are treated with Nrf2 activators or exposed to oxidative stress, Nrf2 must be phosphorylated for nuclear translocation after dissociation from Keap1, a negative regulator of Nrf2, to promote transcriptional activity of Nrf2-dependent genes. As a representative downstream factor of Nrf2, HO-1 can decompose heme into biliverdin, carbon monoxide and free iron. The produced biliverdin is then converted to bilirubin, which has an antioxidant activity (Jenkins & Gouge, 2021; Yu et al., 2018). These findings indicate that discovering substances that can activate the Nrf2/HO-1 axis might be one of the appropriate strategies to counteract cellular damage caused by oxidative stress. Recently, numerous studies have suggested that various natural products have antioxidant properties that can inhibit oxidative damage-mediated apoptosis in myoblasts through activation of the Nrf2/HO-1 signaling (Rui et al., 2022; Wu et al., 2022; Xu et al., 2021; Zhang et al., 2021). Therefore, we investigated whether MEMC could activate Nrf2 and found that the level of p-Nrf2 expression indicating that Nrf2 was activated was clearly upregulated by H2O2 in MEMC-pretreated C2C12 cells. In addition, in MEMC-treated C2C12 cells under oxidative conditions, the activity and the expression of HO-1 was significantly enhanced, while Keap1 expression was decreased. However, the apoptosis blocking and cytoprotective potential by MEMC in H2O2-treated cells was significantly counteracted by ZnPP, an HO-1 inhibitor, suggesting that activation of HO-1 was at least responsible for the blockade of H2O2-induced oxidative damage by MEMC. Our results support previous studies showing that antioxidant activities of various seaweed extracts and their constituents are due to activation of the Nrf2/HO-1 signaling (Jayawardena et al., 2018; Kang et al., 2015; Lekshmi et al., 2019; Niu et al., 2020; Park et al., 2018). Our results are also in good agreement with those of previous studies showing that Nrf2-mediated activation of HO-1 in myoblasts can act as a protective mechanism against apoptosis induced after mitochondrial dysfunction caused by oxidative stimulation (Li et al., 2020; Park et al., 2021; Song et al., 2022).
Taken together, the present results showed that MEMC could alleviate cell cycle arrest, DNA damage, and apoptosis by mitigating H2O2-induced mitochondrial damage as an ROS scavenger in C2C12 myoblasts. In addition, MEMC as an activator of Nrf2 might contribute to the blockade of oxidative damage through activation of HO-1, indicating that MEMC has a high potential application in the maintenance of myoblast function against oxidative damage. However, further studies are required to determine upstream signaling pathways regulating the activity of Nrf2 by MEMC and other antioxidant signals that might be involved in its antioxidant activity.