Introduction
The aquaculture industry is continuously expanding its horizons to meet the demands for products. Various species of fish have been used in aquaculture to fulfill demands for products in various countries. The Serranidae (Perciformes) consisting of five subfamilies and including approximately 522 species belonging to 73 genera are fish of interest to aquaculture. The genus Epinephelus includes 89 species that are recognized as economically important marine fish, more than one third of which are cultured in Asia for domestic consumption and overseas export (Eschmeyer et al., 2010; Mai et al., 2014).
There have been a number of attempts to culture groupers from seed production to mariculture to overcome difficulties associated with insufficient seed supply. In Korea, the kelp grouper (Epinephelus bruneus) and red-spotted grouper (E. akaara) have been successfully induced to mature and produce fertilized eggs (Kang et al., 2020). However, the long time required to grow to marketable size in a temperate region represents an obstacle to their feasible application in aquaculture. There have been attempts to resolve these issues by interspecies hybridization with giant grouper (E. lanceolatus), which is one of the largest species of Serranidae with a rapid growth rate. Advances in hybridization of groupers resulting in the production of new species with intermediate phenotypes have led to concerns about the correct identification of Epinephelus species, which have often been misidentified in aquaculture (Noh, 2020; Rimmer & Glamuzina, 2019).
The red-spotted grouper (E. akaara), also commonly known as Hong Kong grouper, is one of the most commercially valuable marine fish for aquaculture in a number of Asian countries, including China, Taiwan, and Korea (Lee et al., 2020), despite its listing as endangered in the International Union for Conservation of Nature (IUCN) Red List of Threatened Species (Sadovy et al., 2018). The situation has become more complicated as the most expensive grouper species, including kelp grouper (E. bruneus), red-spotted grouper (E. akaara), and seven-band grouper (E. septemfasciatus), have often been misidentified. To overcome such difficulties, which were exacerbated by the generation of hybrid groupers with intermediate phenotypes, it is necessary to develop a rapid method for precise species identification.
A number of advances have been made in the development of molecular markers that are useful for species identification of marine organisms, including catfish, tilapia, salmon, oysters, and shrimp. Two PCR-based genetic analysis methods have been used for species identification, one of which involves amplification-based detection of the target organism with species-specific primers and the other employs amplification of fragments containing variable sequences, such as introns and microsatellites flanked by conserved regions with universal primers (Hebert et al., 2003; Monaghan et al., 2005). In these methods, species can be distinguished by analysis of the sequence or size of the amplified fragments. One strategy to sequence introns is to design primers corresponding to adjacent exon regions and amplify across the intron, i.e., so-called exon-primed intron-crossing (EPIC) markers (Li et al., 2010).
Prolactin (PRL) is a member of the protein hormone family and is involved in diverse biological activities, including water and electrolyte balance, growth, development, reproduction, immunoregulation, among many others (Manzon, 2002). In most euryhaline fish, it is generally accepted that PRL plays a key role in osmoregulation by controlling ion and water balance (Astola et al., 2003; Breves et al., 2020). While a great deal is known about the gene structure and amino acid (aa) sequence of PRL for fish species of interest to aquaculture, relatively little information is available specifically for groupers (Zhang et al., 2004). In this study, we analyzed the structure of the gene encoding PRL from E. akaara and propose the first intron of PRL as an EPIC molecular marker for distinguishing a major grouper species.
Materials and Methods
Groupers belonging to six species were obtained from Chungsol Susan (Muan, Korea): red-spotted grouper (E. akaara, RG), kelp grouper (E. bruneus, KG), giant grouper (E. lanceolatus, GG), seven-band grouper (E. septemfasciatus, SG), and two hybrid species, i.e., giant-red (hGR) and giant-kelp (hGK). Tissues, including the brain with pituitary gland, gill, stomach, intestine, liver, kidney, and muscle, were dissected from RG following anesthesia with 3-amino benzoic acid ethyl ester at a concentration of 150 ppm (Sigma-Aldrich, St. Louis, MO, USA) for 1 min. All samples were frozen in liquid nitrogen and stored at –80°C until use. Genomic DNA was isolated from the gill via phenol/chloroform extraction. Samples of approximately 0.1 g tissue were mixed with 630 µL TNE urea in microcentrifuge tubes containing 20 µL proteinase K and 70 µL 10% sodium dodecyl sulfate followed by incubation at 60°C for 2 h. To the mixture was added 700 µL PCI (phenol:chloloform:isoamyl alcohol = 25:24:1), which was mixed immediately by inverting and then centrifuged at 18,000×g for 5 min. The aqueous phase was mixed with an equal volume of isopropanol. DNA was precipitated by centrifugation at 18,000×g for 5 min, and the DNA pellet was washed with 75% ethanol. The pellet was suspended in 100 µL DNase-free water containing RNase A and incubated at 37°C for 30 min. DNA was stored at –20°C until analysis.
PCR primers (Table 1) were designed based on the conserved region of PRL genes of several fish on GenBank site (Noh et al., 2013b). PCR was performed in 20 µL 1 × HiQ-PCR mixture (GenoTech, Daejeon, Korea) containing 10 mM Tris (pH 9.0), 1.5 mM MgCl2, 40 mM KCl, 1 mM dNTPs with 0.2 µg genomic DNA of RG and 1 µM primer pair (F/R). Each reaction was carried out with an initial denaturation step at 94°C for 4 min, 30 cycles of reactions composed of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 2 min, followed by a final extension step at 72°C for 5 min. Amplified PCR products were analyzed by 1% agarose gel electrophoresis followed by staining with ethidium bromide (0.5 μg/mL). PCR products ligated into an All-in-one™ PCR Cloning Kit (Biofact, Daejeon, Korea) were transformed into E. coli DH5α as described previously (Inoue et al., 1990). Plasmid DNA was isolated using a plasmid purification mini kit (Thermo Fisher Scientific, Seoul, Korea) according to the manufacturer’s protocol. Sequencing was carried out by GenoTech.
The full-length aa sequences of PRL were aligned with other PRL genes obtained from the National Center for Biotechnology Information (NCBI) GenBank database. Phylogenetic trees were constructed using the neighbor-joining method (Saitou & Nei, 1987) with MEGA ver. 5.2. The reliability of each tree node was assessed by bootstrapping with 1,000 replicates. A number-of-differences model was set as the substitution model, and gapped or missing aa residues were treated by pairwise deletion. Aa sequences used to build the phylogenetic tree were obtained for E. coioides (AAR97730), A. schlegelii (AAX21764), A. melanopus (AEB00558), O. niloticus (NP_001266715), C. autumnalis (CAA80660), E. lanceolatus (XP_033500110), Anguilla anguilla (XP_035254403), and O. mykiss (NP_001118205). PRL sequences from Papio hamadryas (ADG56475) and Homo sapiens (AAH88370) were used as outgroups.
To analyze the tissue-specific expression profile of PRL in red-spotted grouper, RNA was isolated from the brain, gill, stomach, intestine, liver, kidney, and muscle using RiboEX™ reagent (Cambio, Cambridge, UK) according to the manufacturer’s protocol. Tissue samples (~0.1 g) homogenized in 1 mL RiboEX™ reagent (Cambio) were mixed with 200 µL chloroform followed by centrifugation at 12,000×g for 10 min at 4°C. The supernatant was mixed with an equal volume of isopropyl alcohol and centrifuged at 12,000×g for 5 min at 4°C. The RNA pellet was washed with 80% ethanol and then treated with RNase-free DNase I at 37°C for 20 min followed by purification on a HybridR column (Tribioscience, Sunnyvale, CA, USA) according to the manufacturer’s protocol. Quality of total RNA was analyzed via 1.8% agarose gel electrophoresis followed by staining with ethidium bromide and quantification using a microvolume nucleic acid spectrophotometer (ASP-2680, V4.1; ACTGene, Piscataway, NJ, USA).
First-strand cDNA was synthesized using an M-MLV cDNA synthesis kit (Enzynomics, Daejeon, Korea) according to the manufacturer’s protocol. The reaction mixture containing 80 μM oligo dT primer and 1 µg total RNA was incubated at 70°C for 5 min and chilled on ice for 5 min. Reverse transcription was performed by adding 20 µL of a mixture containing 10 × M-MLV RT buffer, 2 μM dNTP, 40 U RNase inhibitor, and 200 U M-MLV reverse transcriptase and incubating it at 42°C for 60 min. The reaction was stopped by inactivation at 72°C for 10 min and then the sample was stored at –20°C until use. PCR was carried out to amplify the PRL gene using the primers listed in Table 1 together with a primer set for β-actin from olive flounder (Paralichthys olivaceus) as an internal positive control. The specific primer assays were carried out with pituitary cDNA serial dilutions, showing amplification efficiencies approaching 100%.
PCR was carried out with primers E1F and E2R designed corresponding to the first and second exons of the PRL gene, respectively, and genomic DNA as the template according to the procedure outlined above. The sizes of PCR products were analyzed by 1% agarose gel electrophoresis followed by staining with ethidium bromide.
Sixty red-spotted grouper (8.2 ± 0.1 cm total length, 8.3 ± 0.4 g body weight) were divided into 10 fish in six tanks (0, 4, 8, 24, 48 and 144 h). All fish were adapted to each tank containing seawater (32 psu) before the experiment. With the except of the control group (0 h), the salinity of the experimental tanks was decreased to 8 psu by adding freshwater. Ten fish were sampled at the different exposure times of 0, 4, 8, 24, 48 and 144 h. Fish were dissected after anesthesia by 150 ppm 3-aminobenzoic acid ethyl ester (Sigma-Aldrich) for 1 min to collect brain, gill, stomach, intestine, liver, kidney, and muscle. Collected samples were frozen in liquid nitrogen and stored at –80°C until use.
Results and Discussion
Groupers, including the red-spotted grouper E. akaara, have been of interest for their superior market value as well as growth rate depending on the species. To explore their potential for development as a successful aquaculture species, it is important to understand the characteristics of the key molecules, including PRL, involved in various aspects of their physiology affecting the efficiency of the aquaculture system. To examine the primary structure of PRL in red-spotted grouper, the full-length gene was obtained by PCR amplification and DNA walking of genomic DNA and cDNA templates. The results showed that the gene encoding PRL consisted of five exons of 42, 114, 108, 183, and 189 bp in order from 5′ to 3′, yielding an open reading frame encoding a protein of 212 aa residues composed of a putative signal peptide of 24 aa and a mature protein of 188 aa. The gene included four introns of 1,293, 183, 1,908, and 1,035 bp in order from 5′ to 3′, and all sequences found at the borders of introns contained the consensus GT and AG splicing signals (Fig. 1).

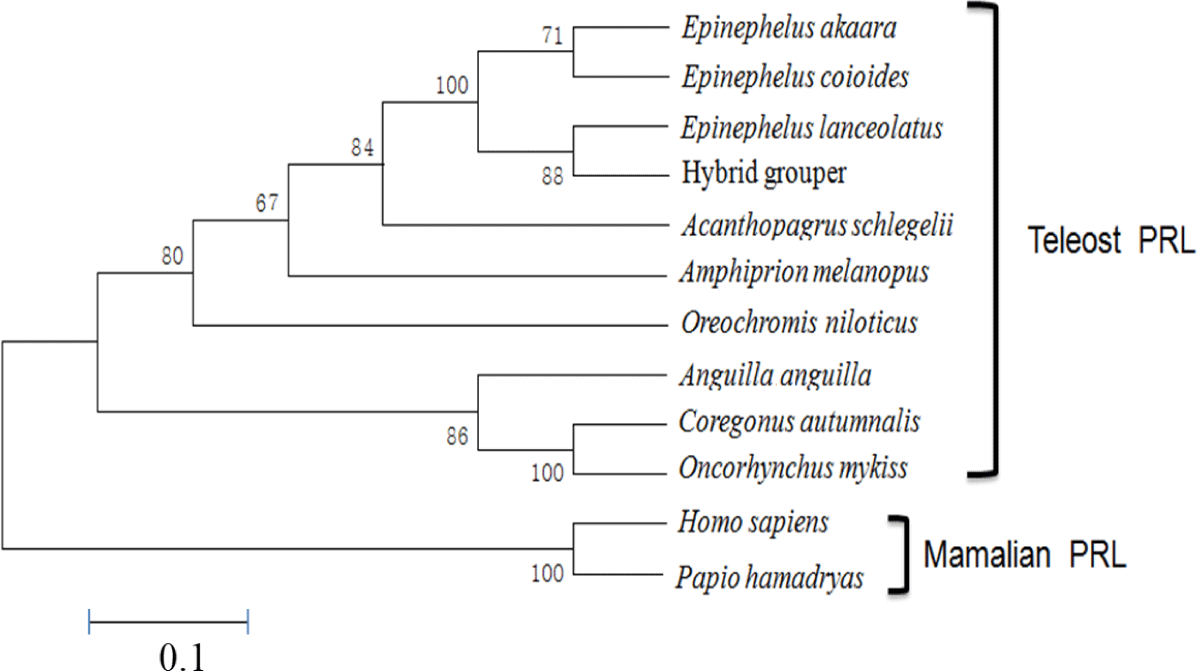
The aa sequence of PRL was deduced from its nucleotide sequence. Comparison of the aa sequence of RG PRL with those of other teleosts was performed using ClustalW and the results indicated an identity of 99% with E. coioides, 83% with A. melanopus, 82% with A. schlegelii, 75% with O. niloticus, 70% with C. autumnalis, and 67% with O. mykiss. RG PRL also contains four cysteine residues in the loci conserved in the PRLs of other teleosts (Fig. 2). Phylogenetic analysis indicated similarity between E. akaara PRL and those of other teleosts but a broad distinction from non-teleost PRLs (Fig. 3).
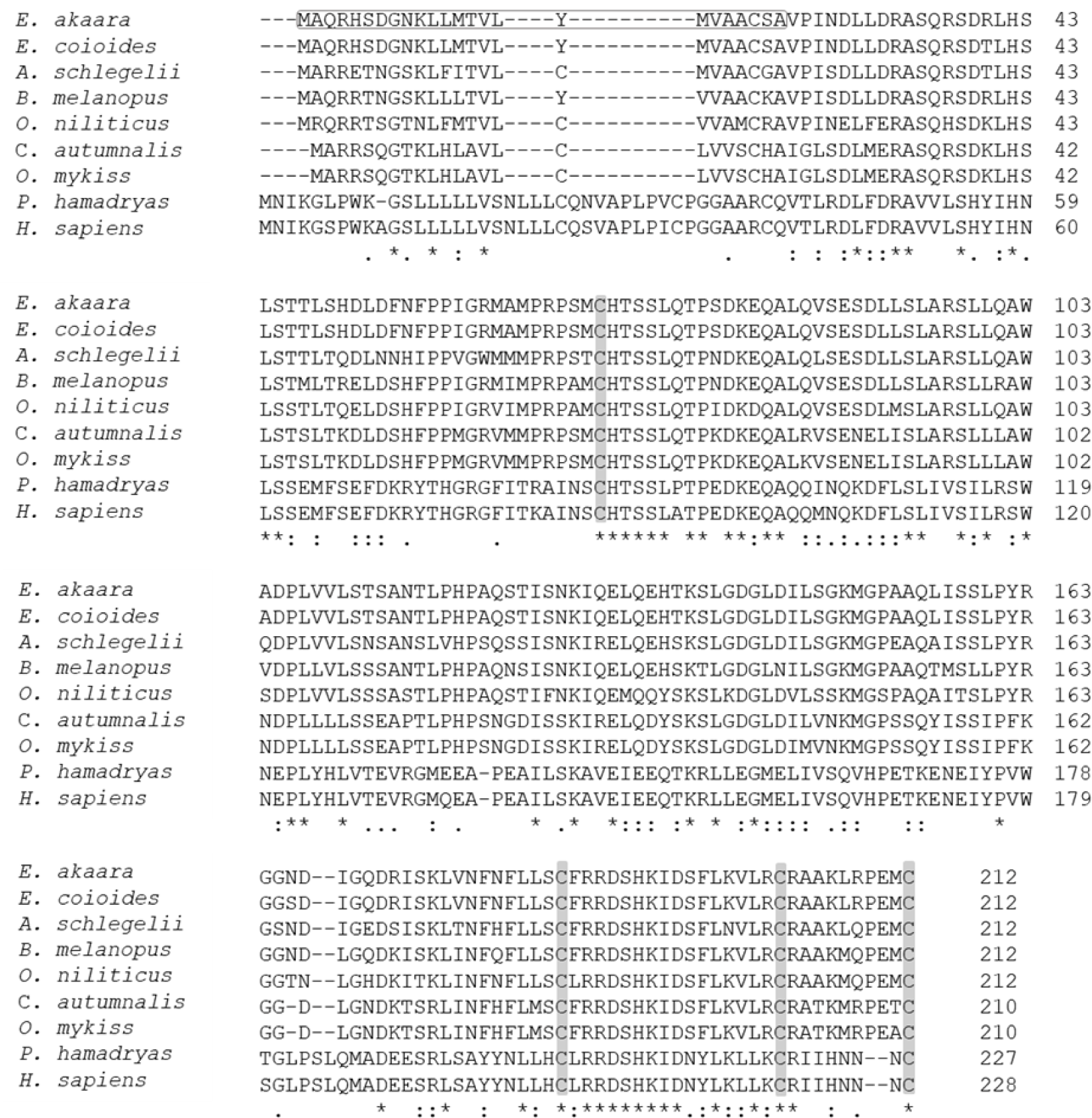
PRL is a multifunctional hormone found in all vertebrates together with PRL-like protein in jawless fish, one of the oldest groups of living vertebrates (Gong et al., 2022). Despite its wide range of functions, all PRL genes share similar structural motifs, including five exons interrupted by four introns with an exceptional exon 1a found in humans. We found that a single form of PRL with a putative signal peptide of 24 aa and a mature protein of 188 aa shared 99% aa sequence identity with that of orange-spotted grouper E. coioides. These structural characteristics are consistent with PRLs reported from other fish, including salmon, tilapia, sea bream, and other groupers (Astola et al., 2003; Swennen et al., 1992; Xiong et al., 1992; Zhang et al., 2004). Overall, teleost PRL was synthesized as a prohormone with a signal peptide of 23–24 aa and full-length aa sequence of varying length depending on fish species (Manzon, 2002): 185 aa in A. anguilla (Quérat et al., 1994), 186 aa in Cyprinus carpio (Chao et al., 1988), 187 aa in Hypophthalmichthys molitrix (Chang et al., 1992), two PRL forms of 188 aa and 177 aa in Oreochromis mossambicus (Yamaguchi et al., 1988), O. niloticus (Rentier-Delrue et al., 1989), and Oncorhynchus keta (Song et al., 1988) in contrast to ovine and human PRLs with 199 aa residues (Cooke et al., 1981; Verma et al., 1989). The presence of four conserved cysteines that may lead to the formation of two disulfide bridges in the C-terminal region is consistent with most teleosts except sturgeon and lungfish, which have three disulfide bonds, similar to mice and humans. Nile tilapia PRL188, which lacks the N-terminal disulfide bridge, appears to be more similar in structure to mouse growth hormone (Manzon, 2002).
RT-PCR was performed to analyze the tissue-specific expression pattern of the PRL gene. The gene was expressed mostly in the brain, including the pituitary gland, of RG, with little if any expression in the other tissues examined (Fig. 4A). A similar pattern of expression was also observed in the hybrid grouper generated between E. akaara × E. lanceolatus (personal observation). These observations are consistent with those in other species, including tiger pufferfish Takifugu rubripes, black porgy A. schlegelii, and starry flounder Platichthys stellatus (Chang et al., 2007; Lee et al., 2006; Noh et al., 2013a). On the other hand, in the orange-spotted grouper E. coioides, goldfish Carassius auratus, and olive flounder P. olivaceus, PRL gene expression has also been detected in other tissues, including the testis, ovary, liver, kidney, spleen, gill, and muscle. This may be due to differences in the level of exclusive dissection of the pituitary resulting in relatively less drastic differences in the level of detection under the conditions tested. However, such a difference could also be plausibly explained by different species of fish in different stages of development in different habitats, considering that there may be more than 300 potential roles of PRL in vertebrates (Si et al., 2021).
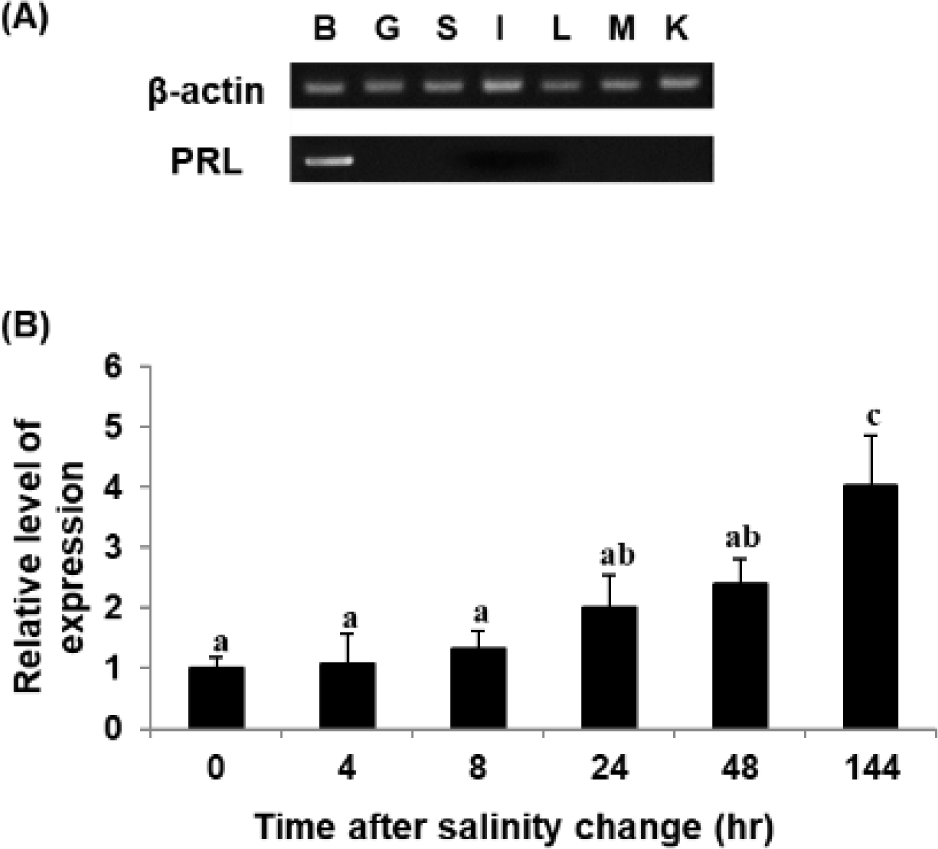
The PRL transcript was also analyzed upon shifting to 8 psu. The level of PRL transcript increased upon exposure of RG to low salinity under the conditions tested (Fig. 4B), consistent with observations in other fish species. However, the experimental fish adapted to 8 psu for 144 h showed the reascent of PRL gene expression, and this fact differs from the results of other fish species (Noh et al., 2013a). This may have been because not all juvenile E. akaara survived under salinity of 8 psu, suggesting continuous stressful conditions and vulnerability to low salinity; this is different from GG, which can survive low-salinity shock (Lim et al., 2016).
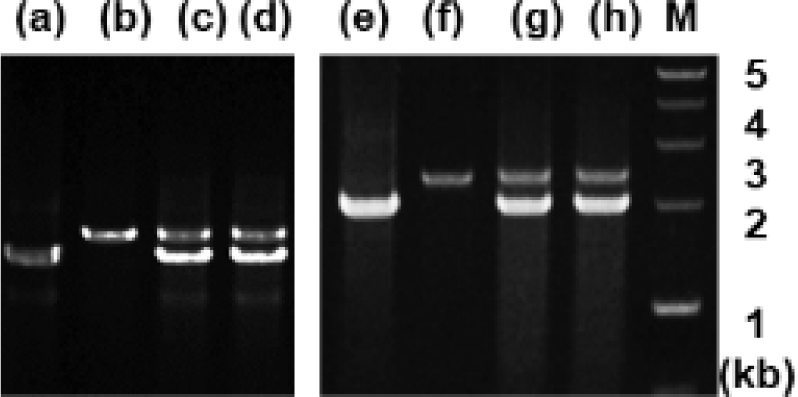
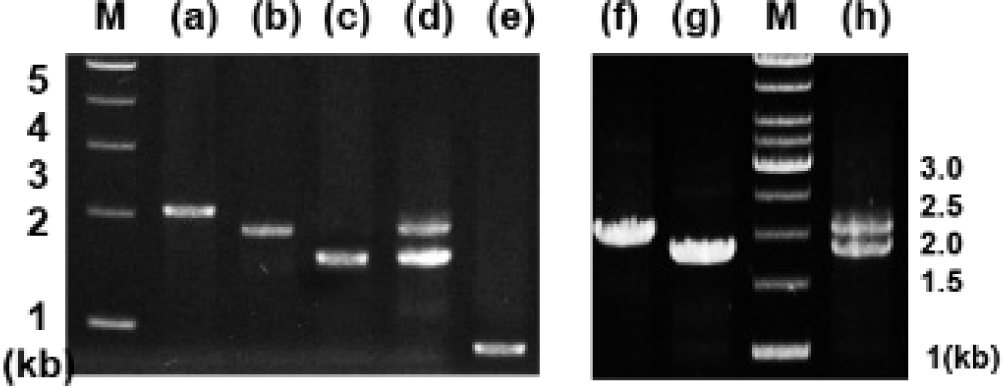
Analysis of genomic DNA and cDNA templates indicated a similar structure of the PRL gene consisting of five exons interrupted by four introns in the grouper species examined. The PRL gene in giant grouper E. lanceolatus also consists of five exons of 43, 113, 108, 183, and 192 bp leading to an open reading frame of 212 aa residues, similar to RG (personal observations). While the overall sizes of the exons in the PRL genes of the two grouper species were similar to each other and to other fishes, the sizes of the four introns in the PRL gene of the giant grouper (1,671, 90, 2,325, 1,277 bp listed from 5′ to 3′) were different from those of RG. To examine whether the differences in intron sizes can be used to distinguish among the species often indiscriminately identified as valuable fish, the sizes of the corresponding regions of the PRL gene in other groupers were compared following amplification. These were first tested with red-spotted grouper, giant grouper, and the hGR hybrid generated between them (Lim et al., 2016) using primers designed based on the conserved sequences of exons of RG to amplify the region encompassing each intron. There were clear differences in the sizes of the amplified fragments encompassing the first intron (Fig. 5) among RG, GG, and the hybrid. In particular, the presence of a double band corresponding to the size of the parental copy indicated its applicability for identifying artificially produced hybrids, some of which have phenotypes that are indistinguishable from the parental species. These results indicate that this method may be useful for distinguishing not only different species but also hybrids generated between them. This study was further extended to other Epinephelus species, including seven-band grouper, and the hybrids produced between giant grouper, which has the fastest growth rate, and other groupers known to have good meat quality. The distinct differences in the sizes of the amplified products (Fig. 6) indicated that most of the commercially important grouper species could be clearly identified by EPIC of the PRL gene. The method is advantageous compared to 16S rRNA-based detection of three species of grouper, i.e., E. bruneus, E. septemfasciatus, and Niphon spinosus (Park et al., 2013), requiring primers for each fish species. Our method may not only facilitate grouper species identification but also help expand the horizons of a rather sporadically used intron-based fish-identification method.