





Introduction
Vibrio vulnificus is an aquatic bacterium infected with ingestion of raw or undercooked seafood or with exposure of wounds to seawater causing gastroenteritis, wound infection, and sepsis. In the case of primary sepsis, the fatality rate reaches more than 50% (Hlady & Klontz, 1996; Oliver, 2005; Shapiro et al., 1998). In South Korea, where seafood intake is high, V. vulnificus has been steadily detected nationwide, and 37 to 88 cases of Vibrio sepsis have been reported every year from 2000 to 2020, and the mortality rate is 45.7% from 2011 to 2020 (Cho & Park, 2019; KDCA, 2021; Kim et al., 1987; 1990; Park et al., 2019). V. vulnificus has several virulence factors (VF) such as capsular polysaccharide (CPS), iron uptake, hemolysin (vvhA), protease (vvpE), repeats-in-toxin (RTX) toxin, lipopolysaccharide, pili, and flagella, and various factors regulating them have been reported for the pathogenicity of V. vulnificus, and studies such as identification of the toxic activation mechanisms have been conducted, too (Jones & Oliver, 2009; Lee et al., 2019).
Along with the development of next-generation sequencing technology, genome analysis of many pathogenic bacteria important in public health has been performed, and in the case of V. vulnificus, starting with the first genome report in 2003, whole genome analysis has been conducted to identify genomic characteristics and various attempts are being made such as comparison between clinical and environmental genotypes (Chen et al., 2003; Morrison et al., 2012; Pan et al., 2017; Pang et al., 2020; Roig et al., 2018). However, compared to other pathogenic bacteria such as Salmonella enterica and Staphylococcus aureus, the genome analysis of V. vulnificus is insignificant, and even compared to the same genus Vibrio, it is less than that of Vibrio parahaemolyticus and Vibrio cholerae (NCBI, 2023).
The eastern coast of Gadeok island is provided good conditions for the habitat of V. vulnificus due to the inflow of the Nakdong river. Additionally, the final treated water from three sewage treatment plants is flowing in this area, and it is possible to influx pollutants from the land. Our previous studies identified the detection tendency of V. vulnificus in the seawater of the eastern coast of Gadeok island, the genetic characteristics related to pathogenicity, and the antibiotic resistance characteristics of the isolates (Oh et al., 2020, 2021). In this study, whole genome sequencing analysis was performed on the pathogenic V. vulnificus isolated from the coast of Gadeok island, and the characteristics of VFs were looked for.
Materials and Methods
V. vulnificus 1908-10 used for whole genome sequencing analysis was isolated in August 2019 from the seawater of the eastern coast of Gadeok island in a previous study (Oh et al., 2020, 2021). This strain possessed virulence-related genes (vvhA, viuB, and vcgC), β-hemolysis activity, and cefoxitin resistance (minimal inhibitory concentration 32 μg/mL). The strain was cultured at 35°C with Luria-Berani broth (NEOGEN, Lancing, MI, USA) supplemented with 1% NaCl for 12 h, and the genomic DNA was extracted using Genomic DNA extraction kit (Bioneer, Daejeon, Korea) according to the manufacturer’s procedure.
Two different genomic DNA libraries were constructed according to the manufacturer’s instructions for the Illumina and the PacBio platform. Sequencing was performed using PacBio Sequel I System (Pacific Biosciences, Menlo Park, CA, USA) and Illumina HiSeqX ten sequencer (Illumina, San Diego, CA, USA). CANU v1.7 (Koren et al., 2017) and Pilon v1.21 (Walker et al., 2014) were used for de novo assembly. The completeness of the genomic data was assessed by B U SCO v5.1.3 (Manni et al., 2021). The genome sequences of V. vulnificus 1980-10 were deposited in the National Center for Biotechnology Information (NCBI) GenBank server under the accession numbers CP118438 and CP118439 for chromosome I and II. Gene annotation was conducted using prokka v1.13 (Seemann, 2014), eggnog 4.5 (Huerta-Cepas et al., 2016) and PATRIC v.3.6.12 (Wattam et al., 2017). The functional classification for coding sequences was performed through Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi) based on the Clusters of Orthologous Genes (COGs) database (2014 update version; http://www.ncbi.nlm.nih.gov/COG/) and was visualized with a circular map using CIRCOS v.0.69-63 (http://circos.ca).
The identity of V. vulnificus 1908-10 was confirmed by comparative phylogenetic analysis using MEGA11 v.11.08 (Tamura et al., 2021) against 16S rRNA sequences of the genus Vibrio. A verage nucleotide identity (ANI) matrix was constructed via EDGAR 3.0 (Dieckmann et al., 2021). The NCBI dataset was used for comparative genomic analysis of V. vulnificus in this study (Table 1).
The VFs of the V. vulnificus 1908-10 genome were analyzed using the virulence factor database (VFDB) 2019 (Liu et al., 2019) and their locations were edited on a visualized circular map using Adobe Photoshop 2023. Virulence gene presence/absence comparison was performed by obtaining comparison strains information from the NCBI dataset and reannotating using prokka. We used PROSITE (Sigrist et al., 2012) to find multifunctional-autoprocessing repeats-in-toxin (MARTX) toxin repeat regions and used MEGA 11 to identify the number and location of repeat sequences described by Roig et al. (2011). NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to perform and compare searches for the effector domains.
The search for antibiotic resistance genes in V. vulnificus 1908-10 was performed using ResFinder (Bortolaia et al., 2020) and Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020).
Results and Discussion
As a result of whole genome analysis, V. vulnificus 1908-10 was composed of two circular contigs, and no plasmid was identified. The total length of the genome was 5,018,425 bp, and its GC content was 46.9%. Contigs 1 and 2 were 3,273,700 bp and 1,744,725 bp long, respectively, with 46.7% and 47.3% corresponding guanine-cytosine (GC) contents. Total coding sequences (CDS) were 4,352, with 2,890 in contig 1 and 1,462 in contig 2. 119 tRNAs and 34 rRNAs were identified and were mainly distributed in contig 1 (Table 2).
As a result of analysis based on the COGs, the genome of V. vulnificus 1908-10 had the most genes related to metabolism at 31.6%, genes related to cell processing and signals at 29.3% and genes related to information storage and process were 16.2%. Mobilome-related genes such as prophages and transposons were 1.8% (Fig. 1). In detail, genes involved in signal transduction mechanisms were the most common at 8.3%, followed by genes involved in transcription at 7.1%, genes involved in amino acid transport and metabolism at 6.8%. Genes involved in cell wall/membrane/envelope biogenesis were 5.7%. The COGs of V. vulnificus Vv180806 and V. vulnificus VV2014DJH have been reported and generally show similar trends. However, in the cytoskeleton category, V. vulnificus Vv180806 and V. vulnificus VV2014DJH had no corresponding genes, while two genes were identified in V. vulnificus 1908-10 showing differences (Pan et al., 2017; Pang et al., 2020).

In a phylogenetic analysis comparing 16s rRNA using MEGA 11 software, this strain was identified as V. vulnificus distinct from other Vibrio spp. and outgroup (Fig. 2 A). When comparing the similarity of genomes using EDGAR based on the NCBI dataset, V. vulnificus 1908-10 was the most similar to V. vulnificus FORC_077, with about 99.41% similarity, followed by V. vulnificus CMCP6 and V. vulnificus FORC_053 was 99.02%. V. vulnificus FORC_017 followed with 98.92%. Strain FORC_077, CMCP6, and FORC_017 were of type C, often found in clinically isolated strains when classified based on genotype. V. vulnificus FORC_053 was of type E. Twelve (80%) of the top 15 isolates with an ANI greater than 98% were type C, so V. vulnificus 1908-10 was similar to type C overall (Fig. 2B). 95 % ANI corresponded to the recommended cut-off point for species delineation (Goris et al., 2007).

As a result of analyzing through VFDB, V. vulnificus 1908-10 had genes of VFs such as mannose-sensitive hemagglutinin type IV pilus, CPS, flagella, metalloproteinase, vibriobactin related to iron absorption, heme receptor, and periplasmic binding ATP binding cassette (ABC) protein transport system. It possessed the luxS gene of autoinducer-2 in the quorum sensing class, and among the secretion systems, the EPS type II secretion system was identified. Toxin factors like vvhA, tlh, and RTX gene clusters (rtxABCD) were identified (Table 3). Genes related to attachment, motility, and secretion systems were located in contig 1 of the genome, while genes for metalloproteinases, iron uptake vibriobactin, transport systems, and toxins such as hemolysin and RTX were identified in contig 2. CPS genes were identified in both contig 1 and contig 2 (Fig. 3). CPS has been reported to be biochemically and genetically diverse among strains (Pettis & Mukerji, 2020), and V. vulnificus 1908-10 was found to have cpsABCDFHIJ, hp1, wbfU, wbfV/wcvB, wbfY, wza, wzb, and wzc (Table 3).
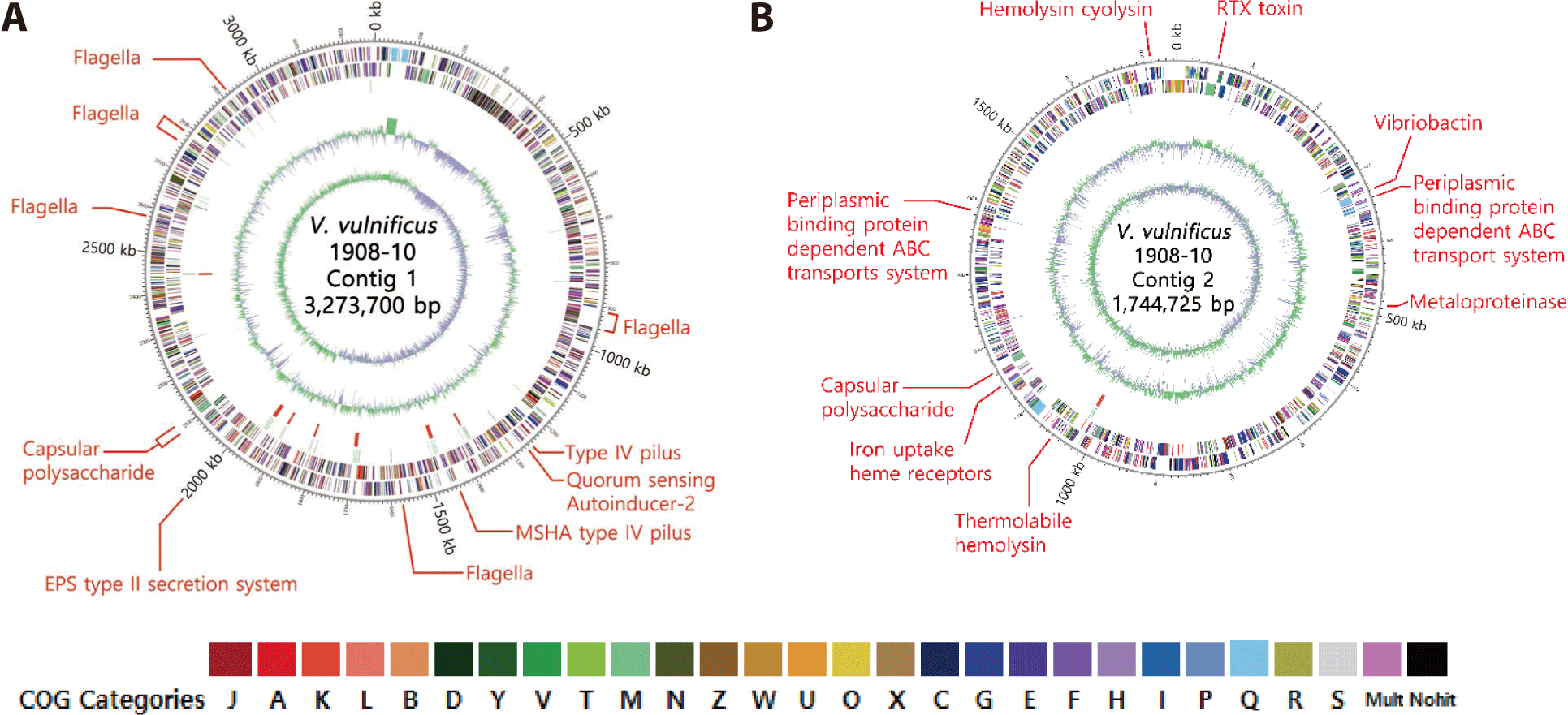
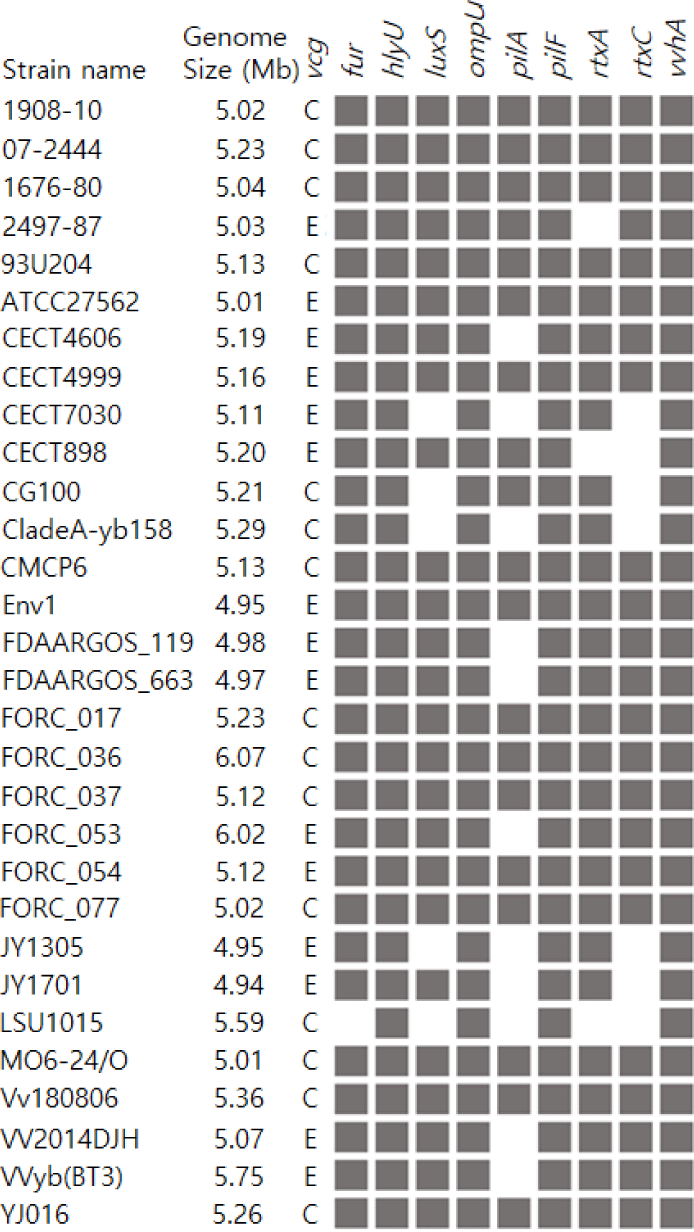
Compared to the 29 V. vulnificus strains in the NCBI dataset, V. vulnificus 1908-10 had fur, hlyU, luxS, ompU, pilA, pilF, rtxA, rtxC, and vvhA genes. All 30 strains including 1908-10, had hlyU, ompU, pilF, and vvhA genes in common. On the other hand, fur was identified in 96.7%, rtxA in 90%, luxS in 83.3%, rtxC in 76.7% and pilA in 60% of the strains. Particularly, pilA, which is part of the type IV pili operon, was found in 80% of type C strains but in 40% of type E strains, showing differences between genotypes (Table 3 and Fig. 4). hlyU regulates the expression of rtxA1 at the transcriptional level, affecting its cytotoxicity and virulence (Liu et al., 2007). ompU is a factor involved in bacterial adhesion to host cells (Goo et al., 2006). pilF is a protein gene required for the assembly of type IV pili, whose functions include surface motility of the strain, colony, and biofilm formation, and host cell adhesion (Alm & Mattick, 1997; Hobbs & Mattick, 1993). vvhA is a hemolysin/cytolysin gene of V. vulnificus.Fur regulates the production of hemolysin, rtxA encodes the RTX toxin and rtxC encodes the toxin activator (Lee et al., 2013; Lin et al., 1999). luxS is an autoinducer-2 synthase gene in the quorum sensing system and has been reported to affect the transcription of vvhA and vvpE (Kim et al., 2003). Overall, V. vulnificus 1908-10 had more similar virulence characteristics to type C strains, as 80% of type C strains had all nine genes compared, while 40% of type E strains did.
MARTX toxins are large single polypeptide toxins produced by various gram-negative bacteria. They deliver numerous effector proteins from the bacteria to the host cell to alter the target cell physiology. In contrast to the conservation of the MARTX toxin domain structure among most of the V. cholerae isolates, the V. vulnificus MARTX toxins are strikingly diverse (Kim, 2018; Satchell, 2015). Ten different effector domains have been recognized among the various MARTX toxins of Vibrio spp., although any one toxin carries only two to five effectors (Satchell, 2015). MARTX toxin of V. vulnificus 1908-10 contained 8 A-type repeats (GXXGXXXXXG), 25 B.1-type repeats (TXVGXGXX), 18 B2-type repeats (GGXGXDXXX), and 7 C-type repeats (GGXGXDXXX). NCBI BLAST showed that the RtxA protein of V. vulnificus 1908-10 had the effector domain in the order of actin cross-liking domain (ACD)-C58_PaToxP-like domain-α/β hydrolase-C58_PaToxP-like domain (Fig. 5), with the same domain sequence as V. vulnificus FORC_017, V. vulnificus FORC_053, and V. vulnificus CECT4999 (NCBI, 2023). Their amino acid sequence of the effector domain was 100% identical to V. vulnificus FORC_017, but there were differences in some amino acids when compared to V. vulnificus FORC_053 and V. vulnificus CECT4999. ACD disrupts the cell cytoskeleton and inhibits the engulfing activity of phagocytic immune cells of the host. α/β hydrolases reduce the intracellular phosphatidylinositol 3-phosphate levels and blocked autophagic/endosomal pathways. The C58_PaToxP-like domain, also known as makes caterpillars floppy-like domain (MCF), is associated with apoptotic cell death (Kim, 2018).
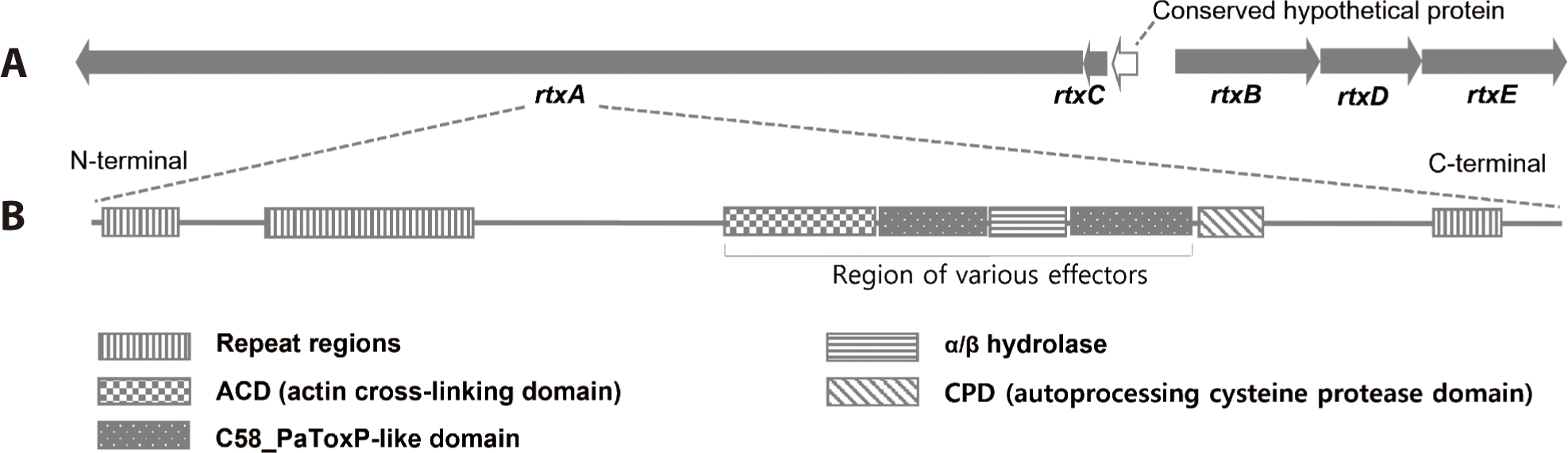
V. vulnificus FORC_077, which had a high ANI value for its genome, had the effector domain sequence of membrane localization domain (MLD)-α/β hydrolase-C58_PaToxP-like domain. V. vulnificus CMCP6 and YJ016 had domains of MLD-α/β hydrolase-C58_PaToxP-like domain–toxin_MLD (toxin effector region membrane localization domain)-RtxA-like domain (C2-2 like domain of various multidomain toxins) (NCBI, 2023). Biochemical mechanisms and direct target molecules for specific effector domains of MARTX toxins have been characterized, but some still require further study (Kim, 2018). Meanwhile, the diversity of effector domains in the MARTX toxin was independent of genotype (data not shown).
Regarding antibiotic resistance in V. vulnificus 1908-10, no acquired resistance genes were identified in the analysis using ResFinder. On the other hand, CARD analysis identified the antibiotic resistance genes crp, adeF, varG, parE, and ftsI in this strain. Of these, ftsI was detected from a protein variant model associated with antibiotic target alteration in cephalosporins, but whether it was a factor of cefoxitin resistance in this strain requires further study (data not shown).
Genome analysis of V. vulnificus 1908-10 isolated from the eastern coast of Gadeok-do r evealed that this strain was similar to C-genotype V. vulnificus in terms of ANI and VFs. MARTX toxin sequence was identified also. V. vulnificus is continuously threatening food hygiene and public health. Among strains that genomes have been reported in South Korea, there has been no report of isolates from riverine seawater. The genomic information of this strain can be used as basic data for the genome of the strain according to the isolation environment as well as understanding the genomic characteristics and virulence of V. vulnificus.