





Introduction
Investigation of the principles and mechanisms determining the geographic distribution of genetic lineages at the intraspecific level, specifically called phylogeography (Avise, 1998), is crucial for the purposes related to the conservation and sustainable use of biodiversity. Analysis of genetic variation within and across populations allows one to reconstruct historical events that have shaped the geographic distribution of the species. Comprehension of genetic structure and interconnectedness among populations also allows one to place conservation priority by recognizing regions that need protection and creating methods to maintain biodiversity (López & Bonasora, 2017). Present-day patterns of the population’s genetic structure are believed to result from a combination of historical events such as climate change and contemporary processes associated with the species’ life history, ecology, and demography (Smith et al., 2011). Some might argue that the phylogeographic patterns of a species could be approached by comparing them to those of sister species co-distributed within the same zone. Depending on the life history traits and environmental conditions, the argument may hold for specific circumstances but not others. While historical and biogeographic events may exert similar effects on different species inhabiting the same zone, species-specific demographic and ecological traits may result in different outcomes, losing their phylogeographic concordance. An intraspecific phylogeographic study on a particular species is essential, mainly when specific interest is in the species understudy. The analysis becomes more critical when it occurs in a geographic zone of high complexity, such as the Sundaland.
The Sundaland was an exposed landmass during the Pleistocene Sea level low stands. It comprised southern Indochina, the Thai-Malay Peninsula, Sumatra, Java, Borneo, the shallow marine shelf (the ‘Sunda Shelf’) between these islands, and West Sulawesi (Hall, 2014). Based on geological history, the Sundaland is a complex zone formed and influenced by several geological events. Geologically, the landmass grew through two major phases: the late Palaeozoic (Permian-Triassic) and the late Mesozoic (Cretaceous) (Hall, 2014).
Concerning biodiversity, Sundaland is regarded as one of four biodiversity hotspots in Southeast Asia, along with Indo-Burma, Wallacea, and the Philippines (de Bruyn et al., 2013). A comparative phylogeographic study across these biodiversity hotspots discovered that Indochina and Borneo have been the source of most biodiversity in the area. Mechanisms by which fish biodiversity flourishes in this area were expected to result from diversification, immigration, and emigration (de Bruyn et al., 2014). The global climate change associated with glaciation during the Pleistocene is thought to have affected its biodiversity and distribution, including freshwater fish. During the period of glaciation, the climatic condition in Southeast Asia was drier and cooler than at present, with longer dry seasons, shorter wet seasons, and more pronounced seasonality (Harrison et al., 2006). The region’s river systems underwent modifications due to Sundaland’s exposure during the Pleistocene. The ancient river systems of the Sunda and Sahul shelves can be seen on maps showing shorelines at various sea levels below the present sea level (Voris, 2000). These river systems were hypothesized to have connected major islands of the Sundaland and facilitated the dispersal of ichthyofauna, as shown by a phylogeographic study with Channa striata (Tan et al., 2012).
The bony-lipped barb, O. vittatus (Valenciennes, 1842), is a freshwater fish found in many locations in Southeast Asia (Kottelat, 2013). It inhabits the rivers and lakes of major Indonesian islands like Java, Sumatra (Syandri, 2014), and Kalimantan (Parenti & Lim, 2005), the Mekong fisheries area of Thailand, Laos, and Myanmar, and the main river systems of Sekong, Sesan, and Srepok in Cambodia, Laos, and Vietnam (Baran et al., 2011). It is a potamodromous fish that completes all its life cycles in freshwater. It lives in smaller rivers, moves to the flooded areas during the onset of the rainy season, returns to its river homes, and sometimes stays in the main river (Riede, 2001).
Considering the geological complexity in the formation and development of Sundaland along with global climate change as well as the biology and life history characteristics of the O. vittatus, the species provides an excellent model to study the comparative forces of historical events and contemporary in shaping the genetic structure of this species across the Sundaland. The present study aimed to explore the phylogeographic structure of the species by using mitochondrial cytochrome oxidase I (mtCOI), a widely used molecular marker for phylogeographic analysis. We hypothesized that the dispersal capacity of this species would lead it to be genetically less structured. However, historical events leading to physical fragmentation, particularly the episodic glacial and interglacial during the Pleistocene, may leave a profound signature in the genetic structure of the populations.
Materials and Methods
The mtCOI DNA sequences used in this study came from two major sources. The primary data originated from samples collected from field surveys, and the secondary data was extracted from the public database (NCBI, 2023). The primary data samples were collected to represent O. vittatus of the main Indonesian islands comprising the Sundaland: Java, Sumatra, and Kalimantan. A few secondary data available on the public database that were qualified to represent the area generated by previous authors were also used. Combining the two sources, 46 O. vittatus mtCOI DNA sequences originating from ten localities were used in this study. A detailed description of the sampling location, sample size, and geographic information are provided in Table 1, and a map illustrating the relative position among sampling localities within the Sundaland is presented in Fig. 1. The geographic location of the samples extracted from the database was traced based on the information provided in the database or related publications. If no specific information was available, the geographic location was approached based on the broader area.
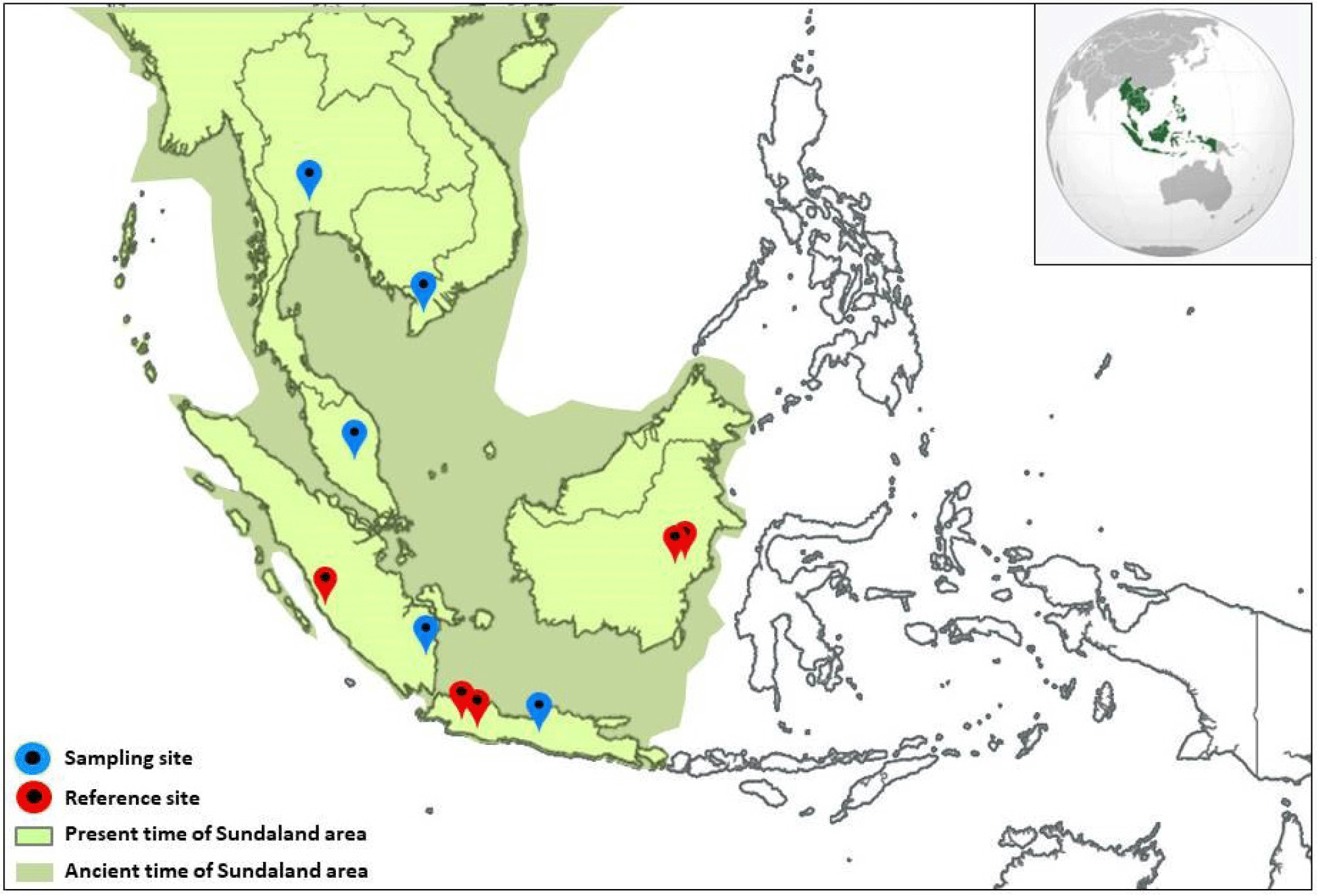
The samples collected from the field study covered five localities: two localities in Java (Bogor and Tasikmalaya), two localities from Kalimantan (Jempang Lake and Melintang Lake), and one location representing Sumatra (Batang Liki, Solok). The laboratory works to generate the mtCOI DNA sequences, including the steps of genomic DNA extraction, PCR amplification, and sequencing. A Brief description of the respective stages is described below.
Each sample’s caudal fin tissue (± 2 cm) was taken using 70% alcohol-sterilized surgical scissors. The fin tissue was dried using a wipe paper tissue and stored in a 1.5 mL microtube containing 1 mL absolute ethanol until the genomic DNA extraction process. If the sample is to be directly extracted, the fins are cleaned first with distilled water, then dried and cut into small pieces before the genomic DNA extraction stage is carried out. The genomic DNA of each sample was extracted using a Genejet Genomic DNA Purification kit (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol.
The mtCOI amplification was performed on a Veriti-96 Well Fast Thermal Cycler (Applied Biosystems, Waltham, MA, USA) PCR system. The PCR amplification was carried out using the MyTaq HS Red Mix Master Mix Bioline Kit with DNA sequence of forward and reverse primers FishF1 (5’TCAACCAACCACAAAGACATTGGCAC3’) and FishR2 (5’TAGACTTCTGGGTGGCCAAAGAATCA3’), respectively. The reaction composition consists of 25 μL PCR master mix, two μL forward primer and two μL reverse primer (20 pmol), four μL DNA templates, and nuclease-free water up to a total volume of 50 μL. The PCR program applied was initial denaturation at 95°C for 3 minutes, followed by 40 cycles of denaturation at 95°C for 30 seconds, annealing at 60°C for 1 minute, and elongation at 72°C for 1 minute. The final extension following the cycle was at 72°C for 5 minutes. To check for the success of amplification, 3 µL of a PCR amplicon of each sample was migrated on a 1.5% electrophoretic gel (30 mL TBE 1×, 0.45 agarose, 3 µL peq green gel dye) together with DNA ladder 100 bp plus (NL1404, Vivantis, Shah Alam, Malaysia) as a marker. The gel was run in an electrophoresis system containing 1/2 TBE at 100 volts for 30 minutes. The electrophoresis results were visualized and documented on the Gel Doc UV transilluminator. The successfully amplified amplicon was then sent to a BRIN sequencing service for Sanger Sequencing.
The corresponding mtCOI sequences from the database were extracted by applying a searching strategy combining query O. vittatus and mtCOI. The NCBI-derived sequences were carefully handled to ensure that the gene regions of interest were comparable with those primary sequences obtained through laboratory work described previously. Based on the nucleotide sequence of a complete mitogenome of Osteochillus hasselti, a sister species of O. vittatus, measuring 16,537 (Accession: NC_029442), the entire length of the Cytochrome oxidase I gene was around 1,500 bp, and its position spanned between 5,500 to 7,000 bp. Aligning the primary sequence data against that genome indicated they were between base positions 5,500–6,200. Each corresponding sequence extracted from the database was aligned to the reference mtCOI genome to ensure they were from the same gene region. The accession number of sequences fulfilling the previously described criteria were KU692714 for Rawa Pening, MN243482 for Lampung, HM156365-HM156368 for Malaysia, MK448104 and MK448073-MK448075 for Thailand, and MW147439-MW147440 for Vietnam (NCBI, 2023). DNA sequence editing and alignment were done using the software GeneStudio Professional Edition version 2.2.0.0 (GeneStudio, Suwanee, GA, USA).
Following the cleanup of the mtCOI sequences, an alignment of 506 base pairs was obtained from all populations and was used for downstream analyses. Analysis of genetic variation and its partition within and between populations was conducted using analysis of molecular variance (AMOVA) implemented in Genalex 6.5 (Peakall & Smouse, 2012). Several genetic variation parameters occurring in within-, between- and whole-population were estimated. For each population, nucleotide diversity (Pi), number of haplotypes (H), and haplotype diversity (Hd) were estimated. Genetic differentiation (Nst) and nucleotide divergence (Dxy) were analyzed for population-pair comparisons. To address how the patterns of genetic variation are distributed over the Sundaland, pairwise comparisons were conducted between populations within and between major localities. The major locality was determined based on the likelihood of geographic isolation leading to a lack of gene flow among the populations. Based on this, six major localities were determined: Java, Sumatra, Kalimantan, Malaysia, Thailand, and Vietnam; from now on, they will be referred to as major localities. Bayesian analysis of population structure (BAPS) (Corrander et al., 2006) has confirmed these six major localities as the most optimal cluster number. The major sampling localities in Indonesia consist of 2–3 localities. This paper uses the term sub-localities for clarity and terminological consistency when referring to this group of localities. Pair-wise comparisons between populations within major localities were conducted for Java, Sumatra, and Kalimantan, where at least two populations occurred with those respective major localities. The first three groups are in Indonesia, while the rest are in different countries. The analyses used DnaSP 6 (Rozas et al., 2017), dedicated software for analyzing DNA sequence polymorphisms. An F-test was conducted to explore partitioning total genetic variation within and between populations. Furthermore, to examine possible factors that contributed to genetic variation, a multiple Mantel test correlating matrices of genetic distance, geographic distance, and a categorical variable for within or between major localities was performed using the software Genalex 6.5 (Peakall & Smouse, 2012).
An individual mtCOI gene tree showing evolutionary relationships among individuals and different populations was reconstructed using the Maximum likelihood method by incorporating a sister species, O. hasselti, as an outgroup. Before performing the analysis, a test to choose the DNA substitution model under bayesian information criterion (BIC) that best fits the data was implemented in MEGA XI (Tamura et al., 2021) using the maximum likelihood method and the following parameter setting: automatic (Neighbour joining) tree, complete deletion for missing data, and no branch swap. Running the procedure returned the Kimura’s (1980) two parameters with invariable sites (K2+I) as the best model. Under this model, the value was 0.13. The parameter values obtained were then used for further related analyses; the phylogenetic trees of individuals were reconstructed using a maximum likelihood. For the maximum likelihood method, the following setting was used. The Kimura’s 2-parameter model with invariant sites (K2+I), complete deletion for missing data, Neighbour-joining for an initial tree, and nearest-neighbor-interchange (NNI) for ML heuristic method were applied. The consistency of phylogeny was tested using a bootstrap method (Felsenstein, 1985) with 1,000 replications. Besides an intraspecific gene tree, a haplotype network showing the evolutionary relationships among haplotypes was created using Popart software (Leigh & Bryant, 2015). The haplotype proportion and distribution over the major sampling locality were overlaid on the map to improve the visualization.
The intraspecific divergence times between pairs of major geographic populations were estimated through a phylogenetic approach by comparing it with an outgroup sister species, O. hasselti. The estimation was carried out using the Bayesian method implemented in a software package BEAST version 1.10.4 (Drummond & Rambaut, 2007). The input file for running in the BEAST was prepared in a BEAUti, a software utility that was part of the BEAST (Drummond et al., 2012). The resulting trees were then summarized for their distribution of various continuous parameters using TRACER version 1.7.1 software (Rambaut et al., 2018). The Tree Annotator version 1.10.4 software (Drummond & Rambaut, 2007) was implemented to produce a maximum credibility clade (MCC) tree. The final step displayed the MCC tree using FigTree software. The estimate of divergence time between O. vittatus populations was carried out by transforming the length of the branch in the MCC tree to an outgroup calibration of divergence time between O. vittatus and O. hasselti reported at 17.8 Mya (Kumar et al., 2022) was used as a reference to estimate the intraspecific divergence times between O. vittatus populations.
Results
Following the alignment of 46 sequences representing ten populations of the Sundaland O. vittatus, each of which was 506 bp in length, 420 (83.0%) sites were monomorphic. In contrast, the remaining 86 (17.0%) sites were polymorphic, 85 of which were parsimony informative sites. The mtCOI sequences of the sample collected in this study were deposited in the Genbank under the following accession numbers: OR494043, OR497767-OR497772 for Bogor, OR51143-51149 for Tasikmalaya, and OR511448-0511459 for Solok populations, OR487412-OR487415 for Jempang, and OR481906, OR481909-OR481911 for Melintang populations. The AMOVA revealed that total genetic variation was partitioned into the proportion between population (69%) and within population (31%). Several population genetic parameters of interest in this study, including nucleotide and haplotype diversities and genetic differentiations, are described in Table 1.
A contrasting pattern of nucleotide diversity (Pi) characterized their distribution in the overall and individual populations. A high Pi value (0.04561) was observed at the whole population level. This number is about 40 times higher than those observed within Java, Kalimantan, and Malaysia populations, ranging from 0.000 to 0.001, 25 folds more elevated than Sumatra, and about 20 folds higher than those found in the Thailand and Vietnam populations.
A similar pattern was observed for haplotype diversity (Hd), in which the total Hd = 0.87 was observed in the whole population. The distribution of this diversity among populations was highly variable, ranging from zero to 100%. Several populations, such as sub-localities in Java and Sumatra, were genetically highly homogenous, and only one haplotype was discovered within the respective populations, leading to zero diversity. On the other hand, the Vietnam population showed an extremely high Hd. It should be noted, however, that two different haplotypes were identified from only two samples. Increasing the sample size would probably change this population’s Hd profile. The contrasting profile of genetic diversity distribution between the low in within-population and the extremely high in the whole-population implied that a significant proportion of genetic variation was contributed by between-populations component, such as shown by the parameters of nucleotide divergence (Dxy) and sequence-based genetic differentiation (Nst).
Overall, the pairwise Dxy between major localities over the Sundaland ranged from 1.86% between Java and Sumatra to 12.24% between Vietnam and Malaysia. Within the Indonesian region, the highest Dxy was observed between Sumatra and Kalimantan (Dxy = 3.6%). The divergence level of these Indonesian major localities was comparable to that between Thailand and Vietnam (Dxy = 4%). However, they are only half or even less compared with those outside of Indonesia (Malaysia, Thailand, and Vietnam), resulting in a divergence ranging from 8% to 10%. The Dxy other population pairs were substantial, ranging from 11%–12%. In line with the Dxy, a similar pattern was also observed in genetic differentiation expressed in the Nst. At the broader geographic scale, the pairwise Nst among six major geographic localities ranged from 92% to 99%, showing the pattern of nearly complete genetic differentiation. An extreme contrast pattern occurred within the Kalimantan populations (Nst = 0.033%), showing a lack of genetic differentiation.
In the investigation of possible factors resulting in the observed genetic structure, the multiple Mantel test identified that 76% of the genetic structure was caused by geographic isolation. The actual physical distance explained 43%, while historical events leading to the isolation contributed about 33% of the observed genetic structure.
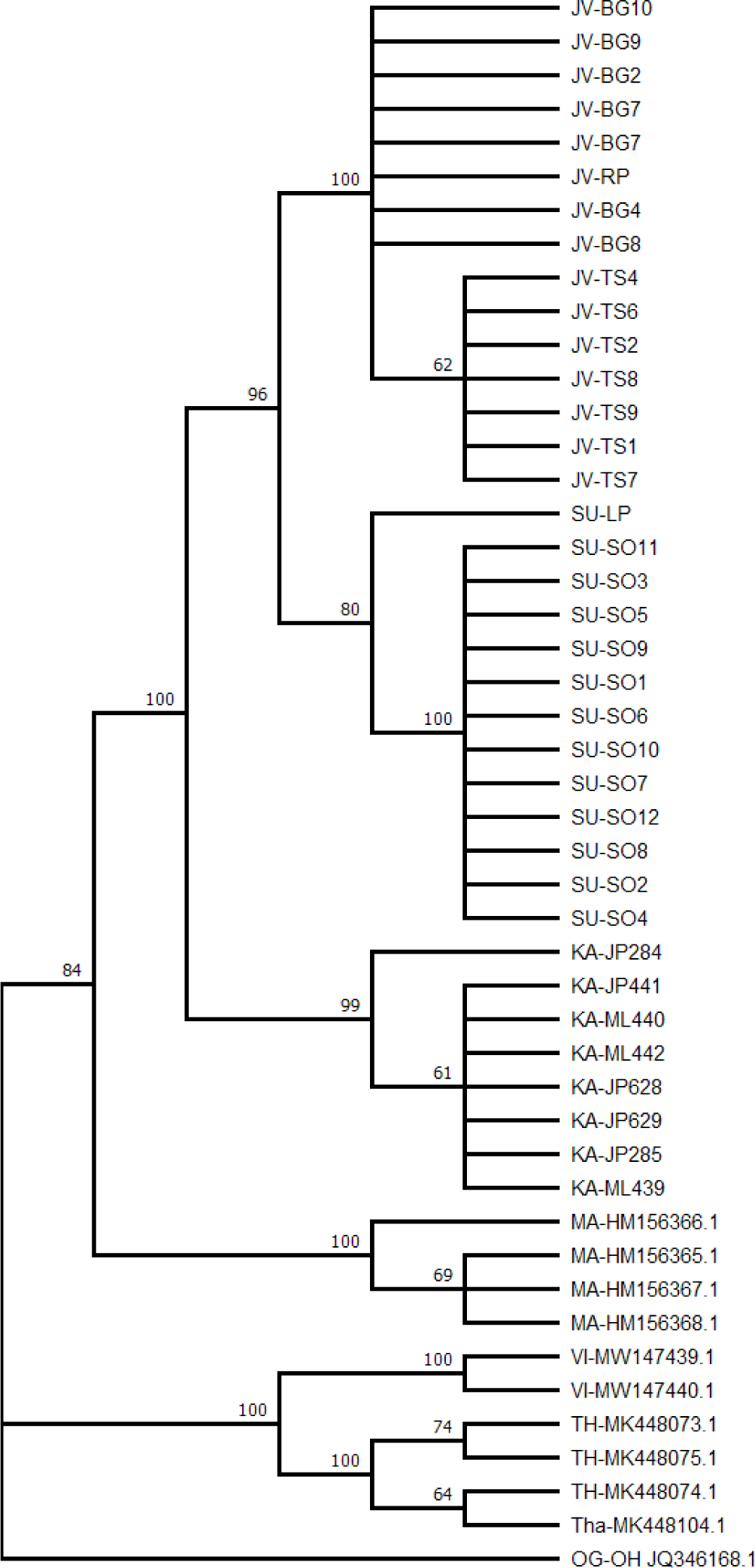
A phylogenetic tree showing the evolutionary relationships among individuals is presented in Fig. 2. Referring to major localities, the tree shows monophyletic clades with strong bootstrap values, ranging from 80% to 100%. Starting from the lower-level clade, the Java and Sumatra populations were evolutionary closer and formed a monophyletic clade. The Java-Sumatra clade formed a monophyletic clade to Kalimantan populations, forming an Indonesian clade. The Indonesian clade formed a monophyly to Malaysian populations, forming Sunda shelf localities. The highest-level monophyletic clade was formed between the Sunda shelf clade and the Indochina populations of Vietnam and Northern Thailand. In addition, within major localities, monophyletic clades were also observed in Java, between Bogor and Tasikmalaya populations, and in Sumatra, between Solok and Lampung populations. The evolutionary relationships among haplotypes and their distribution across sampling localities are presented in Figs. 3 and 4.
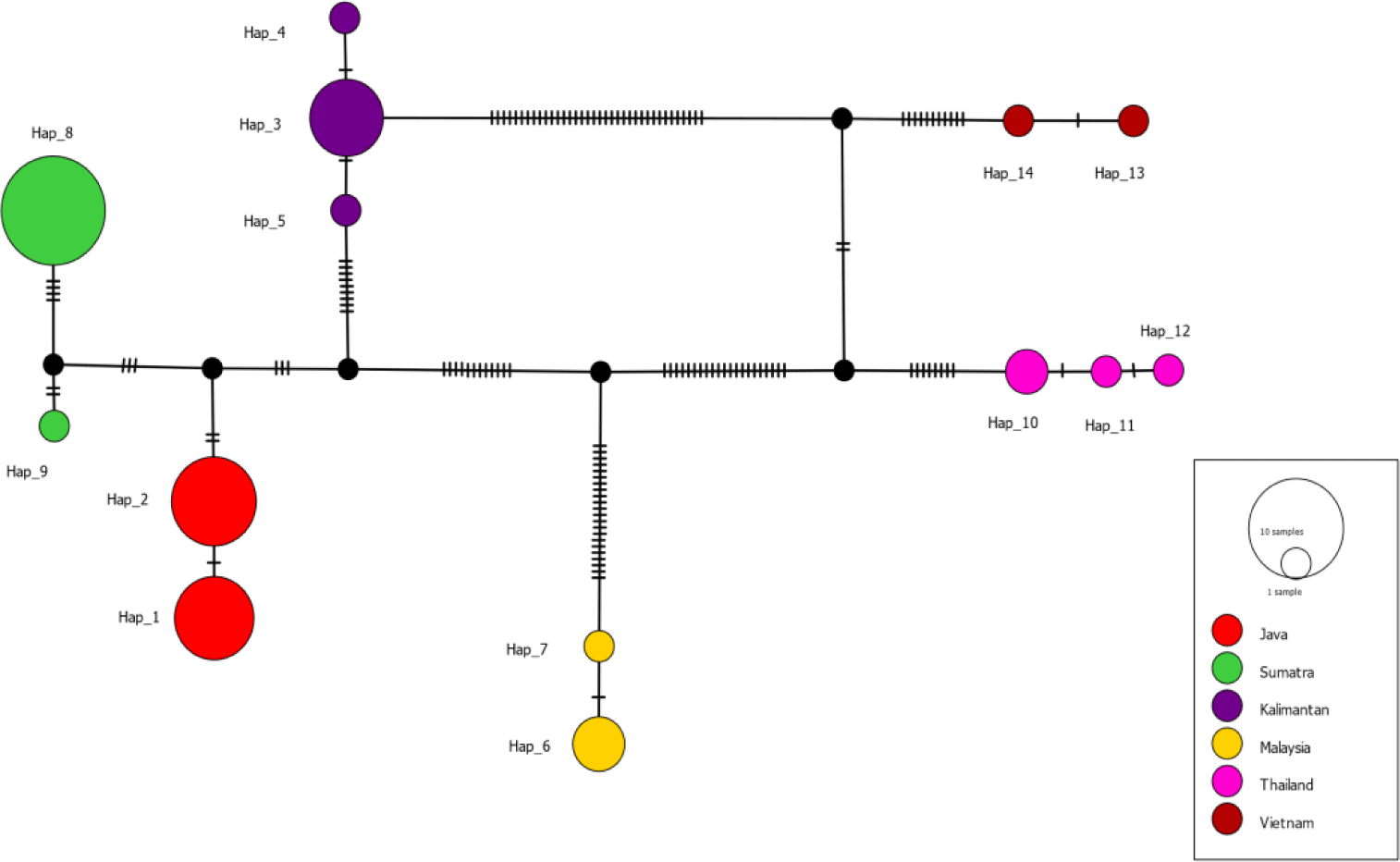
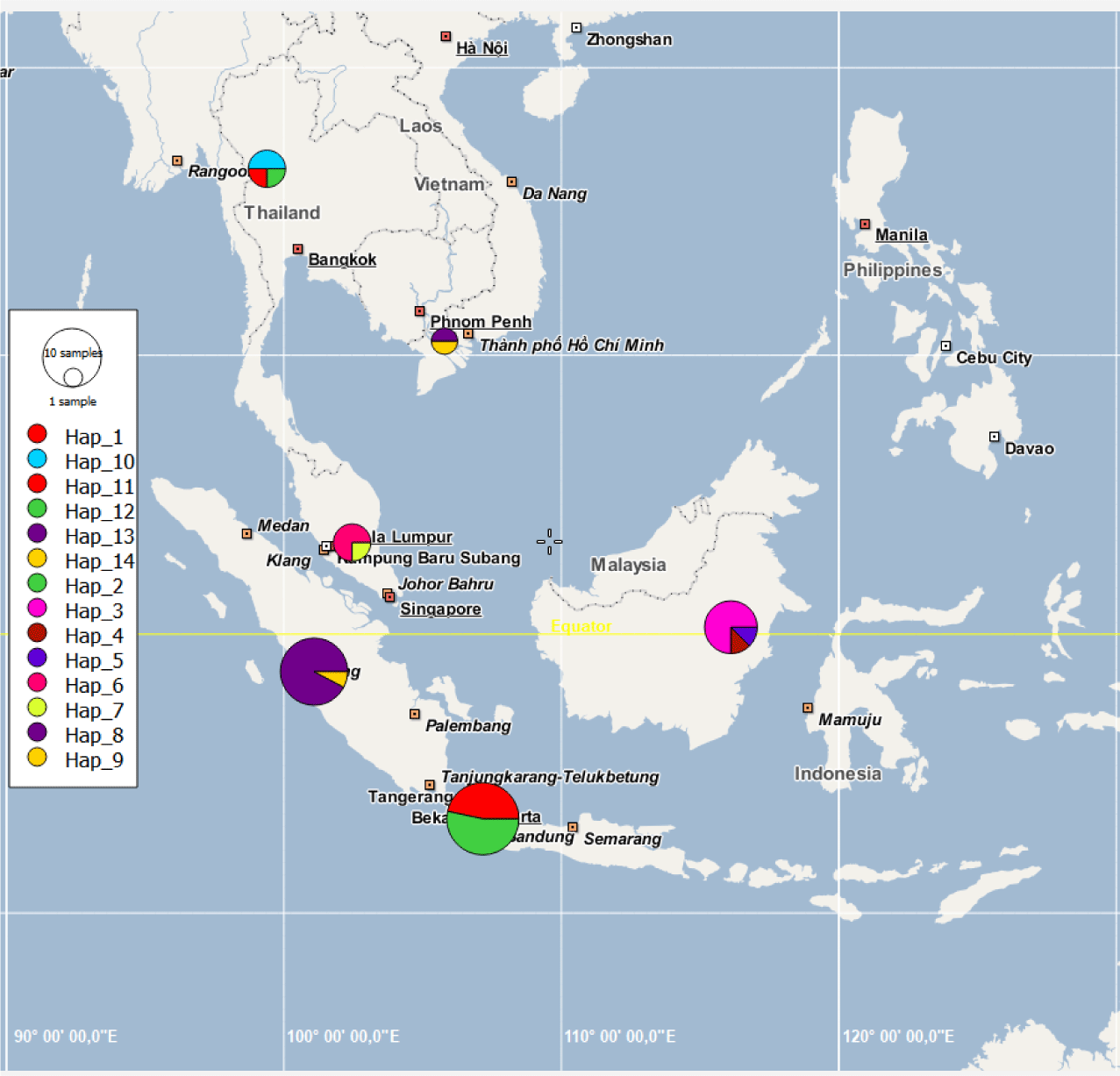
The haplotype network (Fig. 3) shows the haplotypes, with highly variable mutational steps, connected one to another. The number of mutational steps among haplotypes was minimal within the major localities, ranging from 1 to 4 steps, but highly significant between localities, ranging from 5 to 56 steps. Fig. 4 displays the haplotypes extracted from segregating sites and their frequency distribution in six major localities, namely Java, Sumatra, and Kalimantan islands, Malaysia, Thailand, and Vietnam. Based on these, the haplotypes are unique in that no haplotypes are shared among these localities. For instance, haplotypes 1 and 2 were exclusively distributed in Java Island. Neither such haplotypes occurred in other Indonesian islands of Sumatra and Kalimantan, nor Malaysia, Thailand and Vietnam. A similar distribution pattern was observed with other haplotypes within the respective major localities. In contrast to the major localities, the evidence of shared haplotype was observed in sub-localities occurring in Kalimantan and Java Islands. The Kalimantan samples comprising two sub-localities, the Jempang Lake and Melintang Lake, shared one haplotype in common (Hap-3). However, the same case was not applied to Sumatra populations, which, despite quite a large number of samples collected in Solok, no common haplotype between sub-localities Solok and Lampung was observed.
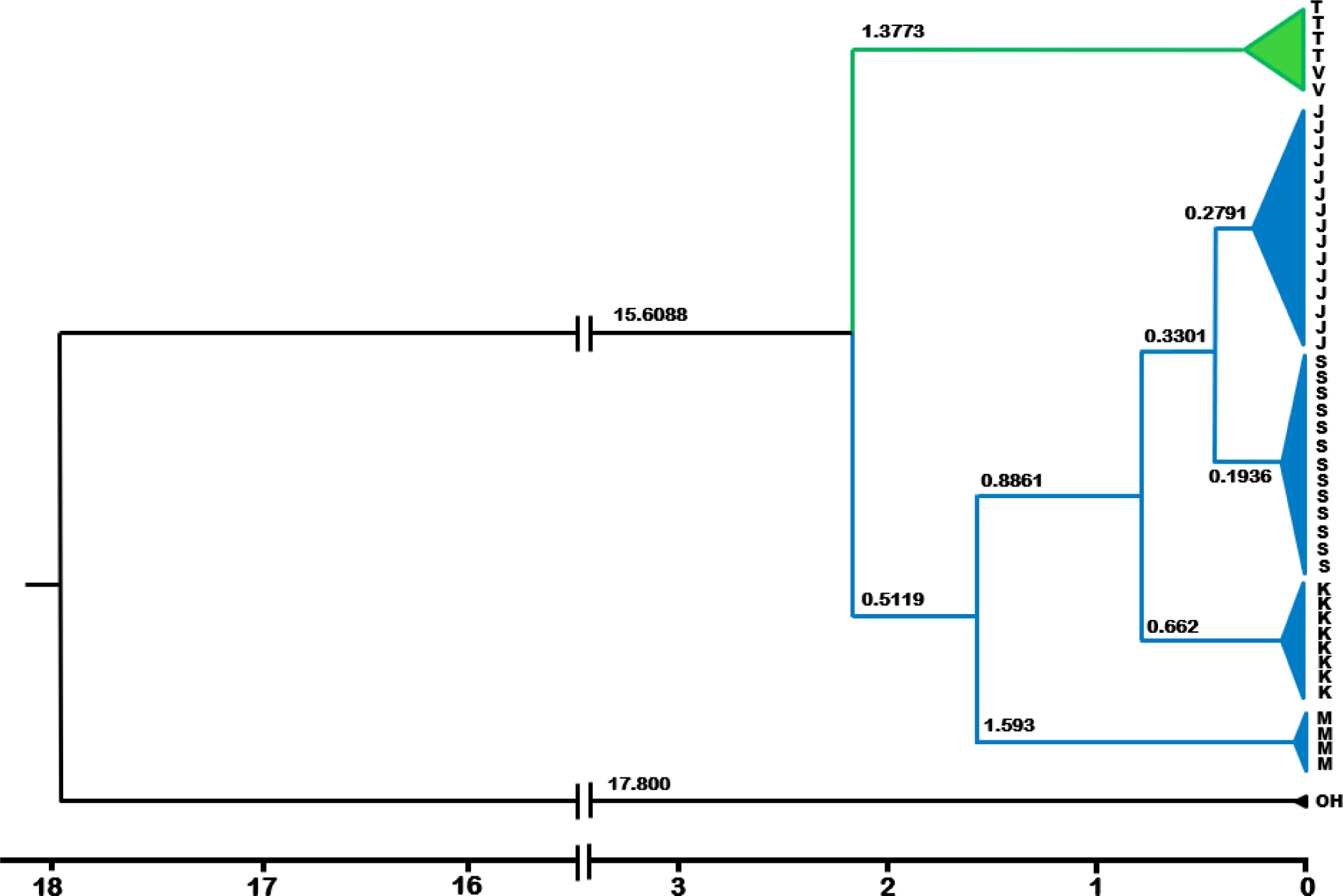
The estimated divergence times between and within major localities is presented in Fig. 5. Two major clades, Indochina (Vietnam and Thailand) and Sunda shelf clades, consisting of Malaysia and Indonesian populations, split one from another at about 2.2 Mya. The split of Malaysia and Indonesia lineages is estimated to have occurred some 1.6 Mya. The Thailand and Vietnam lineages diverged at 0.78 Mya, similar to that between the Kalimantan and Sumatra-Java lineages. The Sumatra and Java lineages diverged some 0.45 Mya, representing the youngest divergence times among major localities (island- or country-based populations. Looking closer at within-Indonesian islands, the most recently diverged populations were between the Jempang and Melintang lakes in Kalimantan at 0.15 Mya, followed by those between Tasikmalaya and Bogor Populations in Java Island, estimated at 0.21 Mya. The most evolutionary diverged populations within-island were between the Solok and Lampung lineages, estimated at 0.29 Mya.
Discussion
Several notable features emerged from the present study. Firstly, there was low genetic diversity within individual populations but extremely high between major localities. Secondly, the intraspecific phylogenetic tree and haplotype network revealed a distinct geographic pattern in haplotype distribution. Thirdly, interpopulation-splitting events were estimated to occur within the Pleistocene. Finally, the analyses showed that the present genetic structure has been mainly formed by geographic isolation resulting from physical geographic distance and historical events, which presented barriers to gene flow.
The significance of genetic variation within a species has been thoroughly acknowledged due to its potential for long-term survival, adaptation to changing environments, and raw material for selection. Currently, the threat to the genetic diversity of aquatic species has been increasing due to anthropogenic activities and global climate, resulting in temperature rise and its corresponding impacts. High genetic diversity is often associated with a better potential for a species to be more resilient and vice versa (Allendorf et al., 2014; Bernatchez, 2016).
The present study showed low genetic diversity and high differentiation among bony lip barb populations over major localities in Sundaland. A similar pattern was also reported in a study with Salmonid in Mongolia (Kaus et al., 2019) and Hexaplex trunculus, a marine gastropod, in the Iberian Peninsula. This pattern, in general, is associated with factors such as the founder effect, genetic drift, and a lack of gene flow due to long-term geographic isolation (González-Tizón et al., 2008). While all aspects are likely, long-term geographic isolation resulting in a lack of habitat connectivity and, thus, gene flow seemed to be the most plausible explanation. Based on historical events, the six major sampling localities, which included the islands of Java, Sumatra, and Kalimantan in Indonesia, the Malaysian peninsula, Thailand, and Vietnam, have been separated by rising sea levels at least since the last glacial period, some 12 Kya. Acknowledging that the glacial-interglacial period had occurred episodically since early Pleistocene 2.6 Mya (Elias, 2013), it could be expected the separation between them has happened since then. The sea level rise has disconnected land bridges that previously connected areas in the Sundaland. During isolation, populations experience independent and probably, to some extent, adaptive evolution, resulting in an accumulated genetic difference.
In this study, the long-term isolation and lack of gene flow leading to the nearly complete genetic are supported by the parameter Nst ranging from 92%–99%. These Nst values indicate an almost complete genetic differentiation resulting from prolonged isolation. In brief, the distribution pattern of genetic variation among major localities of Sundaland is relatively homogenous, characterized by the lack of gene flow and nearly complete genetic differentiation. A similar finding to this study was reported with species of Nematocharax, a characid fish native to Neotropical freshwater ecoregion (Barreto et al., 2022). The patterns of low diversity within the population but high between populations have been reported with European grayling, Thymallus thymallus (Koskinen et al., 2002), and coastal cutthroat trout, Oncorhynchus clarkii (Wofford et al., 2005).
High genetic differences between O. vittatus populations from major localities may indicate that they represent different species or evolutionary significant unit. The possibility that they are different species requires supporting studies of relevant aspects such as biology, morphology, or other aspects according to the species concept that will be used. The concept of biological species, for example, requires verification of their ability to interbreed, which is beyond the scope of this study. Therefore, based on the existing data and information, these populations may be considered as different evolutionary significant units according to their geographic origin (Java, Sumatra, Kalimantan, Malaysia, Thailand and Vietnam). Several other studies reported the similar phenomenon, where there was a high intraspecific genetic distance while maintaining their species integrity. For example, Dahruddin et al. (2017) reported genetic distances ranging 0%–12.81% in populations of Dusky sleeper fish, Eleotris fusca, in Java-Bali while Adamson et al. (2010) reported an average genetic distance of 7% in snakehead, C. striata, in Southeast Asia and India. Understanding and recognizing these geographically isolated populations as distinct evolutionary significant units is important and can have practical implications for fisheries resource management. For example, it will provide information to management authorities not to undertake conservation efforts to restore stocks by translocating and stocking fish from other populations.
The pattern of genetic structure occurring in sub-localities was more heterogeneous. On one end, a high genetic differentiation, leading to nearly complete fixation, was observed between the Bogor and Tasikmalaya populations. Conversely, a less structured and high gene flow, leading to almost pan-mixing populations, was observed between the Jempang and Melintang populations in Kalimantan. Considering these sub-localities’ landscape and the species’ dispersal capacity, the observed patterns were expected. While Bogor and Tasikmalaya are located on the same island of Java, no habitat connectivity and gene flow exist. The population within the respective sub-localities probably experienced the founder effect, genetic drift, or adaptive selection, leading to low genetic diversity, evidenced by monomorphic haplotype, a pattern that was also reported with C. striata populations in the study area part of the Sundaland (Tan et al., 2012). In contrast, the Jempang and Melintang Lakes from which the Kalimantan samples were collected are close to the Mahakam River. While both lakes are about 19 km apart, they receive the same water flowing from the Mahakam River. Additionally, there is also a waterway connecting both lakes, providing a kind of habitat connectivity. In brief, the landscape of the two lakes and the migratory capacity of this species allow them to disperse for either food or reproduction. The role of habitat connectivity in facilitating gene flow has been reported in many aquatic taxa, such as Salmon (Kaus et al., 2019), and snakehead (Tan et al., 2012).
A deep phylogeographical structure can be identified from at least four genealogical concordances: within-locus genealogical concordance, multi-locus concordance, multi-species concordance, and multiple lines of empirical concordances. A within-locus concordance refers to a condition in which several nucleotide alterations along a DNA sequence reliably separate one array of haplotypes from another. Multi-locus concordance refers to the situation in which multiple gene trees consistently place a set of populations into the same groups (Avise, 2009). While we acknowledge the presence of a sample size issue in several localities, the present study revealed a clear and deep genetic structure in O. vittatus populations across Sundaland. The distribution patterns of DNA polymorphisms led to easily identified lineages were observed. The intraspecific maximum likelihood phylogeny (Fig. 2), Bayesian tree topology (Fig. 5), and haplotype network (Fig. 3) clearly show geographic-based sequence clustering, where populations clustered according to the islands and the country of origin. Similarly, sequence clustering based on locality also occurred within a finer geographic scale. The data discovered in this study perfectly matched with the type-1 concordance (Avise, 2009).
Several factors, such as geological features and species dispersal capacity, may drive the formation of phylogeographic break. Among these, species’ life history traits, such as dispersal capacity, and historical events, such as sea level changes affecting habitat connectivity, are highly relevant. O. vittatus is a potamodromous freshwater fish with high dispersal potential (Poulsen & Valbo-Jørgensen, 2000; Riede, 2001) that provided their habitats connected; less structure would be expected. This pattern was the case with the populations of sub-localities Jempang and Melintang, where habitat connectivity has facilitated dispersal and gene flow among them. However, a phylogeographic break would emerge when barriers to dispersal exist and last for an extended period. The present study revealed the latter case with the genetic structure of O. vittatus populations among major localities: Java, Sumatra, Kalimantan, Malaysia, Thailand, and Vietnam. The phylogeographic break due to the barrier to dispersal has been reported for many aquatic taxa separated by the Panama isthmus, separating the Caribbean and Pacific oceans (Avise, 2009).
Phylogeographic patterns have often been associated with historical events, namely Pleistocene sea level changes. Additionally, within the Sundaland, the phylogeographic patterns for freshwater species have also been associated with ancient river systems (de Bruyn et al., 2013; Song et al., 2013; Tan et al., 2012). It is interesting to compare the order of temporal pattern of molecular divergence shown in the phylogeny with the historical regression of Sundaland land bridge exposure associated with sea level changes (Voris, 2000). In the present study, the oldest molecular divergence time, between the Indochina populations and the rest, followed the land bridge pattern loss in Thailand Gulf, separating the Indochina region against Indonesia and Malaysia due to rising sea levels at 50 m BPL. At this contour depth, the land bridge still connected all the Indonesian main islands and Malaysia. At 40 m BPL, the land bridge connecting the western part of a half Malaysian Peninsula to the eastern part of northern Sumatra was lost, while all main Indonesian islands were still attached. Finally, at 30 m BPL, when the land bridge connecting Kalimantan and Sumatra disappeared, Java and Sumatra still connected. This may explain why the split in this clade becomes the youngest in the phylogeny. The land bridge lost in Sundaland impacted the dispersal and distribution of freshwater fishes living in the ancient river systems.
Four ancient river systems are identified in Sundaland: the Malacca Strait, Siam, Northern Sunda, and Eastern River systems (Voris, 2000). The extant populations own some level of genealogical correlation with those in the past, which inhabit those river systems. Under the arrangement of Palaeo river systems, the O. vittatus populations of Indochina may associate with Siam, Malaysia with Malacca Strait, Sumatra with Northern Sunda, Java and Kalimantan with Eastern Sunda river system. Interestingly, clustering based on ancient river systems provides a better explanation of clustering in the phylogeny in a particular situation. This is the case with Solok, Lampung, and Malaysia populations. Based on geographic distance, the distance of Solok-Lampung is similar to Solok-Malaysia, about 600 km. Interestingly, the sequence divergence in the first (1.2%) is much lower than in the second (9.2%). In addition to being located on different islands, different ancient river systems also ran within those islands, so both populations might have distant ancestors. The inclusion of old river systems to explain genetic structure in Sundaland has been reported with halfbeak fish, Dermogeny (de Bruyn et al., 2013), and Channidae (Tan et al., 2012).
The estimated divergence times between O. vittatus populations on major localities in the Sundaland have occurred within the quaternary period that lasted some 2.6 Mya (Elias, 2013). The youngest was 0.45 Mya between the Java and Sumatra lineages, and the oldest was 2.2 Mya between the Vietnam and Thailand clades against the rest of the remaining populations. We acknowledged the possible reliability issue resulting from the use a single calibration point due to lack of available resources. To cope with this, we used the widely referred estimates of closely related species, O. hasselti (Kumar et al., 2022). During the quaternary period, the climate on Earth experienced a repeated oscillation between glaciation and interglaciation intervals, the duration of each on average 100,000 and 10,000 years, respectively (Elias, 2013). During the glaciation period, the earth experienced a cold and dry climate, forming glaciers on continents. A considerable amount of water was locked in these glaciers, resulting in a lowered global sea level and creating land bridges connecting landmasses previously unconnected. The global climate was warmer during interglaciation, and the sea level rose, resulting in the separation. The cycle of glaciation and interglaciation has been thought to affect the distribution and evolution of life on Earth (Johnson et al., 2017).
In comparison with other studies working with different taxa, it is of interest to note the effect of ancient climate, especially the ice age, on the spatial distribution and evolution of ichthyofauna in the region, such as Channidae (Adamson et al., 2010; Tan et al., 2012), and the halfbeak, Dermogenys (de Bruyn et al., 2013). Within the snakehead, C. striata, the intraspecific divergence times between the populations of the mainland, which included Thailand, Lao PRD, and Vietnam, and that of Sumatra Island was estimated at 3.72 Mya. Inferred from the same chronogram tree (Adamson et al., 2010), the time to the most recent common ancestor among populations within the Southeast Asia mainland was estimated at the early Pleistocene, some 2.6 Mya. Within C. micropeltes, a younger divergence time, about 1.2 Mya, was observed between populations of Thailand and Vietnam (Adamson et al., 2010). A more detailed comparative study on the divergence times of ichthyofauna over the Southeast Asia, which also covered the Sundaland, was a study with the halfbeak fish Dermogenys (de Bruyn et al., 2013), covering three biodiversity hotspots in Southeast Asia. The divergence time between Bogor and Bandung (located in West Java) is estimated at 0.8 Mya. The divergence time between the Dermogeny populations of Bandung and Bogor in West Java versus Surabaya in East Java, estimated at 1.2 Mya. A study of phylogeography with Dermogeny taxa across four biodiversity hotspots in Southeast Asia revealed that the patterns of genetic structure within this taxon were best explained by the tectonics and paleo drainage system. The present study, showing some concordance between the estimated population splitting time and the patterns of land bridge loss associated with sea level rise, provides evidence of the historical events in structuring the genetics of populations in Sundaland. This is also consistent with multiple Mantle test results inferring that the historical events leading to barriers to gene flow explained 76% of the presently observed genetic structure in Sundaland O. vittatus.
Conclusion
The bony lip barb in Sundaland shows low genetic diversity within populations but high differentiation between them. Genetic structure patterns were observed in major islands and countries and finer sub-localities lacking habitat connectivity. Historical events, species dispersal capacity, and habitat connectivity have contributed to genetic structure within the species. Although the species is currently not a conservation concern, this study provides baseline information for managing genetic resources conservation and sustainable use in the future.